Keywords
|
| Silymarin; Solubility; Dissolution; Physical mixture; Kneading |
Introduction
|
| Oral absorption of drugs with solubilities <0.1 mg/mL is likely to be dissolution limited. On the other hand, if a drug is too water soluble (and/or too hydrophilic) the dissolved drug molecule will have little tendency to partition from the aqueous exterior into a lipophilic biomembranes (e.g., the eye cornea or gastrointestinal mucosa) and then to permeate the membrane. Thus all drugs must possess some degree of aqueous solubility to be pharmacologically active, and most drugs need to be lipophilic to be able to permeate biological membranes via passive diffusion [1]. |
| For ensuring sufficient bioavailability of drugs in humans, the extent of solubility and speed of dissolution are relevant. Thus, solubility is a key parameter in pharmaceutical research and development as poor solubility is one of the main causes for poor bioavailability [2]. |
| Silymarin (SILY) chosen in the present study is a poor watersoluble drug with oral absorption of about 23-47%, leading to poor bioavailability in-vivo. Presently, a lot of research is being done on bioavailability enhancement techniques of this wonderful molecule from Mother Nature as reported in literature [3,4]. |
| A formulation technique that increases the apparent aqueous solubility and enhances its absorption through biological membranes is one of the most appropriate techniques. One such approach is to formulate/incorporate/disperse a water-insoluble drug into a watersoluble carrier to improve its dissolution in-vitro and absorption invivo [5]. Of late, few attempts to enhance solubility and/or dissolution of silymarin via use of diverse carriers like Water-soluble carriers: β-cyclodextrins [6-8] and PEGs [9] and PVP K30 [10], Amphiphilic carriers: Gelucires 44/14 [11] and Lipid carriers: Phospholipids [12-14] that have led to a highly bioavailable product are reported in the literature. |
| Fulvic acid (FA) is a stable, water soluble carrier moiety, characterized by having a sponge like structure punctured by voids of about 200-1000 Å in diameter, and a number average molecular weight of about 700-2500 [15]. Earlier it has been used as a carrier for poor bioavailable drugs such as Furosemide, Carbamazapine (by humic acid), and Itraconazole in order to increase their bioavailability by its great complexing ability [16-18]. Thus, the aim of our study was to complex SILY with FA to study the effect of fulvic acid as solubilizer of silymarin. Besides investigating the role of FA in improvement of solubility, its effect on dissolution and permeation of silymarin was also studied. The physical mixture and kneading methods were employed and complexes were characterised extensively by DSC and FT-IR spectroscopy. |
Materials and Methods
|
| Silymarin 70% dried powdered extract was obtained as a gift sample from Maneesh Pharmaceuticals Ltd., Mumbai, India and Fulvic acid was extracted in the laboratory by microwave assisted extraction from shilajit capsules. All other reagents and chemicals were of analytical grade. The equipments: Dissolution apparatus (DS 8000, Labindia Pvt. Ltd, India), UV Spectrophotometer, (Shimadzu 1601, Japan), Humidity chamber (Thermolab, India), Electronic balance (Scientific Systems, India), double distilled water, etc. were used during the study. |
|
Phase-solubility study
|
| Phase-solubility studies were carried out at room temperature according to the method reported by Higuchi and Connors [19]. Excess amount of SILY was added to distilled water containing various concentrations of FA (0.2-2% w/v) in stopper conical flask (10 mL) and the resulting mixture was equilibrated in a thermostatic shaking water bath for 48 h on a rotary flask shaker. The suspension so obtained were passed through a membrane filter (0.45 μm) to remove undissolved solid particles and the filtrate was suitably diluted and analyzed spectrophotometrically (Shimadzu, UV 1601) at 288 nm against blanks prepared using the same concentration of fulvic acid in distilled water. |
| From the solubility data, the apparent stability constant (Ka) and Gibbs free energy transfer (ΔG°tr) were calculated with formulas as described by Ahuja et al. in the literature [20]. |
| Apparent stability constant (Ka) of the complexes was calculated from the phase-solubility diagrams according to the following equation: |
 |
| where; |
| So is the solubility of drug at RT in the absence of carrier/ligand and slope means the corresponding slope of the phase-solubility diagrams, that is, the slope of the drug molar concentration versus carrier/ligand molar concentration graph. |
| Values of Gibbs free energy of transfer, ΔG°tr, of SILY from plain water to aqueous solutions of the carrier fulvic acid were calculated according to the following relationship: |
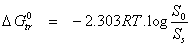 |
| where; |
| So and Ss are the molar solubilities of SILY in 1% w/v aqueous solution of the fulvic acid carrier and in the plain water respectively. |
|
Preparation of Silymarin-Fulvic acid binary systems
|
| Physical mixture: Physical mixture of SILY and FA were prepared by grinding the appropriate amount of mixture for a period of 60 minutes in a clean dry glass pestle and mortar and the resulting mass was passed through a 100 mesh sieve to obtain a uniformly sized powder. The homogenous mixture so obtained was stored in an air tight vial in a dessicator [21,22]. |
| Kneading: Weighed appropriate amounts of SILY and FA were triturated for 15 min a dry clean glass mortar and pestle. During the process, the water content of the paste was empirically adjusted by ethanol and triturated to maintain the consistency of the paste. Trituration was continued until the product started drying on the walls of the mortar. The product was further dried in the hot air oven at 60°C for 15 min, powdered, passed through 100 mesh sieve, transferred in an air tight vial and stored in a dessicator [21,22]. |
|
Characterization parameters
|
| Drug content estimation: The quantities of binary mixtures (equivalent to 5 mg of SILY) were dissolved in water. Appropriate dilutions were made and drug content was measured spectrophotometrically at λmax 288 nm. |
| Aqueous solubility determination: Excess amount of complex was kept in an amber colored bottle containing 10 mL of distilled water and stirred using a thermostatic mechanical shaker (25°C) for 72 hr [18]. Appropriate dilutions were made and drug solubility was measured spectrophotometrically at λmax 288 nm. |
| Differential scanning calorimetry: The DSC thermograms of SILY and FA alone and their binary systems prepared by both the methods were obtained using a Pyris- 6 DSC equipment using aluminium crimp cells with about 2 mg of samples, under dynamic N2 atmosphere (flow rate: 20 mL/min) and at a heating rate of 10°C/min in temperature range from 30-300°C and analyzed [18,21,22]. |
| Fourier transform infrared spectroscopy: The FT-IR spectrograms of SILY and FA alone and their binary systems prepared by both the methods were obtained using FT-IR Perkin Elmer equipment. Pellets of the samples were prepared after grinding and dispersing the powder in micronized IR grade KBr powder in a mortar and pestle, and scanned over a wave number range of 4000-400 cm-1 as reported in literature [18,21,22]. |
| Dissolution study: Dissolution of both the binary systems (equivalent to 25 mg of SILY) was carried out using the USP rotating paddle dissolution apparatus (DS 8000, Labindia Pvt. Ltd, India). The dissolution media was 900 mL double distilled water, pH 6.5 at 37°C, with stirring speed of 100 rpm and paddle depth of 25 mm. Dissolution studies were performed on pure drug (25 mg) and the binary systems containing an equivalent amount of the drug. Aliquots of 5 mL were withdrawn periodically and were replaced with 5 mL of fresh dissolution medium up to 120 minutes. The aliquots were analyzed spectrophotometrically at 288 nm. The percentage drug release versus time graph was obtained and enhancement in dissolution with respect to pure drug was calculated [10]. |
| Ex vivo rat gut sac permeation studies: To determine the effect of complexation on permeability of SILY, non-everted rat gut sac method was employed and compared with that of plain SILY after modification of the method as described in literature [23]. Here, a 5 cm long noneverted intestinal tissue sac basically ileum was taken and thoroughly flushed with 0.9% normal saline to remove any debris from inside. The tissue was mounted on a double jacketed glass assembly containing 50 mL of tyrode buffer solution under controlled temperature 37 ± 2°C with continuous aeration and stirring. The tissue was filled with 1 mL of plain drug dispersion or complexed drug (equivalent to 10 mg of SILY) by both the methods. The samples were withdrawn from outside the sac in intervals, filtered with syringe filters (0.5 μm) and analyzed spectrophotometrically at λmax 288 nm. |
Results and Discussion
|
|
Solubility and phase solubility-interpretations
|
| Solubility is an important parameter that affects the absorption and bioavailability of drugs. Solubility of SILY in plain water at 25°C was found to be 50 μg/mL indicating it as practically insoluble in water [24]. The phase-solubility studies were carried out at room temperature to determine the stoichiometric proportion of SILY with FA. According to Higuchi and Connors there can be five types of phase-solubility relationship [19]. A-type phase-solubility profiles are obtained when the solubility of the substrate (i.e., drug) increases with increasing ligand (carrier) concentration. These are further of 3 types: AL-type, AP- type and AN-type. B-type phase-solubility profiles indicate formation of complexes with limited solubility in the aqueous complexation medium. These are further of two types: BS- and BI-types as shown in Figure 1. From the color of the solutions in the vials (Figure 2), it was quite clear ostensibly that silymarin solubility increased linearly in different fulvic acid concentrations as compared with plain water and these solutions were analyzed spectrophotometrically at 288 nm as shown in Table 1. The SILY solubility increased from 50 μg/mL (in plain water) upto 765 μg/mL (in 2% w/v fulvic acid solution) which was approximately 15 times higher when compared to solubility of SILY in plain water, attributable to the great complexing ability of FA, increased wettability and micellar solubilization of SILY with hydrophilic groups on the water side and hydrophobic nucleus inside the core. In aqueous solutions carriers like FA are able to form inclusion complexes with many drugs by taking up drug molecule or more frequently some lipophilic moiety of the molecule, into the central cavity. The hydrophilic carrier interacts with drug molecules mainly by electrostatic forces and occasionally by other types of forces like hydrogen bonds. No covalent bonds are formed or broken during the complex formation, and drug molecules in the complex are in rapid equilibrium with free molecules in the solution. |
| For the stoichiometric ratio determination of drug and carrier, a graph was plotted between solubility of SILY (μg/mL) versus FA concentrations (μg/mL) (Figure 3). According to Higuchi and Connors, the straight line curve- characteristics of AL type was obtained with r2 value (0.9845) and from this the ratio between the drug SILY and carrier FA was adjusted as 1:1 molar ratio. |
| Apparent stability constant: The value of apparent stability constant, Ka, was computed for 1:1 drug- carrier interaction, since the curve obtained in the present study was AL type with resultant slope less than unity. From the graph, the slope of the straight line was found to be 0.034879 (3.48 × 10-2) and the apparent stability constant (Ka) for silymarin was found to be: |
 |
| Ka=0.00072279(7.22 × 10-4 K-1) |
| High values of Ka obtained for SILY-FA binary solutions revealed strong binding affinity between drug and solubilizer. |
| Gibbs free energy transfer: The solubility values were also used to calculate the thermodynamic parameter of interaction between SILY and FA as shown in Table 2. The Gibbs free energy transfer values were positive at low FA concentration i.e., 0.2% w/v and became negative with increasing FA concentration i.e., 2% w/v demonstrating that the reaction conditions became more favorable as the concentration of FA increased. The negative values indicated the favorable and spontaneous nature of dispersing/solubilizing drug into the carrier system [25,26]. |
|
Characterization of Silymarin and Fulvic acid binary systems
|
| Drug content: The binary systems of SILY-FA were made in the molar ratio of 1:1 w/w by physical mixture (PM) and kneading (KD) technique. The complexes were found to be slightly brown and free flowing powders. The drug content as estimated in both the types of inclusion complexes was found to be approximately 99% for PM and 98% KD. It means SILY-FA complexes made by both PM and KD technique lead to formation of their binary systems. |
| Aqueous solubility determination: Results depicted that complex formation greatly enhanced the aqueous solubility of silymarin and better performance in case of PM (20 times) was seen as compared to KD (17.8 times). |
| DSC: The DSC thermograms of SILY, FA and their corresponding PM and KD binary systems at 1:1 drug: carrier weight ratios are shown in Figure 4. A broad endothermic event between 50°C and 120°C was observed for pure drug SILY. This peak corresponds to melting peak of silybin, an active constituent of silymarin complex [24]. In the thermograms of PM and KD, the drug peaks disappeared in almost both of them but more significantly in PM, this shows that silymarin might be in amorphous state in binary systems. |
| FT-IR: FT-IR is often used to investigate interactions between drug and carrier in binary systems. The spectra of SILY, FA and their corresponding PM and KD binary systems at 1:1 drug: carrier weight ratios were obtained as shown in Figure 5. For FA a broad band around 3400 cm-1 attributed to stretch of hydrogen bonded OH group from phenol or alcohol and/or to those from the carboxylic groups, amide and amine N-H stretching was observed. Peaks at 2925 and 2850 cm-1 can be caused by symmetric and asymmetric stretching vibration of aliphatic C-H bonds present in CH3 and CH2 groups. Apart from these 1540, 1360, 1170, 1080 cm-1 were also prominent [27]. For the pure drug SILY spectrum has been shown only from 2000 cm-1 to 400 cm-1 for clear understanding of peaks in fingerprint region. The drug characteristic peaks were as follows: 1639, 1512, 1464, 1362, 1274, 1162, 1128, 1085, 1030, 995, 821, 781, 739 and 645 cm-1 [24]. In the PM binary system, most of the drug characteristic peaks either got shortened/ diminished or disappeared indicating dispersing of drug in carrier system effectively. In KD binary system also many of the drug peaks got shortened and diminished indicating interaction between the drug and carrier. However the dispersion was found to be better in PM than KD as shown in Figure 5. |
| Dissolution studies: To evaluate the drug release of SILY from PM and KD, complexed SILY was compared with plain uncomplexed SILY (Figure 6). The dissolution rate of uncomplexed SILY was limited, which can be attributed to the poor solubility of SILY in plain water (pH 6.5) with a total of approximately 6.03% only in T5 minutes and upto 13.5% drug release in T30 minutes. The SILY-FA PM at the ratio of 1:1 w/w, showed increase in dissolution rate with 50.5% and 85.27% drug release in T5 and T30 minutes respectively. The SILY-FA KD at the ratio 1:1 w/w also showed increase in dissolution profile with 45.2% and 80.1% in T5 and T30 minutes respectively. In both the PM and KD cases the plateau was achieved at T60 minutes. Increase in dissolution rates was achieved for PM up to 6.13 folds and for KD up to 5.93 folds when compared with plain silymarin powder at the T30 minutes as shown in Table 3. It suggests that the increase in dissolution rate of PM compared to KD is attributed to the formation of better amorphous phase between drug and carrier and this was also seen in solid state analysis studies also where PM showed slightly better results as compared to KD. Although the dissolution profiles of KD did not differ much from that of PM. Thus from here it can be concluded that fulvic acid enhanced the dissolution profile of silymarin significantly. |
| Gut sac permeation studies: The permeability studies for uncomplexed plain SILY and PM and KD were carried out by noneverted rat gut sac method (Table 4). Permeability was very slow in case of plain SILY, whereas both the PM and KD binary systems showed an increase in permeation rate upto 3.69 and 3.29 folds when compared to uncomplexed SILY in T60 minutes as shown in Figure 7. |
Conclusion
|
| Silymarin is a water insoluble drug and shows poor dissolution characteristics. In light of the above fact, inclusion complexes of silymarin with fulvic acid a water soluble carrier were attempted. The phase solubility studies indicated AL type of graph and from this the SILY-FA ratio was fixed as 1:1. The inclusion complexes were prepared by PM and KD methods and were characterized for drug content and aqueous solubility. The PM method showed better drug content and aqueous solubility profiles as compared to KD method. In the solid state characterization by DSC, complete disapparence of drug peak was seen in PM type as compared to KD type. In FT-IR also, drug peaks got diminished more in PM method as compared to KD method. Lastly, dissolution studies and permeation studies were carried out for both the PM and KD binary mixtures. In dissolution studies, 13.5%, 85.27% and 80.1% drug release at T30 minutes was acheived for plain SILY, PM and KD binary systems respectively indicating the enhancement in dissolution of SILY when complexed with FA either by PM or KD methods. An increase in permeation rate upto 3.69 and 3.29 folds was observed for PM and KD respectively when compared to uncomplexed SILY in T60 minutes. Thus, solubility, dissolution and permeability were found to increase when SILY was complexed with FA by either methods, establishing the application/role of fulvic acid as a bioenhancer of silymarin. |
Acknowledgements
|
| Javed S wishes to thank the Indian Council of Medical Research, New Delhi, India for providing financial assistance in the form of Senior Research Fellowship. Maneesh Pharmaceuticals Pvt. Ltd. India for providing the Silymarin 70% extract and Jamia Hamdard for the scientific environment and ambience for carrying out the studies. |
Tables at a glance
|
 |
|
|
|
| Table 1 |
Table 2 |
Table 3 |
Table 4 |
|
Figures at a glance
|
|
 |
 |
 |
 |
| Figure 1 |
Figure 2 |
Figure 3 |
Figure 4 |
 |
 |
 |
| Figure 5 |
Figure 6 |
Figure 7 |
|


