Emilio Franco-Macías1* Liliana Villarreal-Pérez1 Aida Suárez-González2 Irene Pérez-Ortega1 Florinda Roldán-Lora3 and Eloy Rivas-Infante4
1Department of Neurology, University Hospital Virgen del Rocio, Seville, Spain
2Dementia Research Centre, Department of Neurodegeneration, UCL Institute of Neurology, University College London, London, UK
3Department of Radiology, University Hospital Virgen del Rocio, Seville, Spain
4Department of Neuropathology, University Hospital Virgen del Rocio, Seville, Spain
*Corresponding Author:
Emilio Franco-Macías
Department of Neurology
University Hospital Virgen del Rocío
Av. Manuel Siurot s/n. 41013, Seville, Spain
Tel: (+34) 955012593
Fax: (+34) 955012597
E-mail: efranco17@gmail.com
Citation: Franco-Macías E, Villarreal- Pérez L, Suárez-González A, et al. Sporadic Creutzfeld-Jakob Disease: A Four-Year Evolution Case with Heterozygosity at Codon 129 and Kuru Plaques. J Neurol Neurosci. 2016, 6:4.
Received Date: November 25, 2015; Accepted Date: December 04; 2015; Published Date: December 08, 2015
Keywords
Sporadic Creutzfeldt-Jakob disease; Magnetic resonance imaging; Long- duration, Kuru-type plaques; Heterozygosis methionine-valine
Introduction
Sporadic Creutzfeldt-Jakob disease (sCJD) is characterised by rapidly progressing dementia, myoclonus and extrapyramidal and pyramidal involvement. Patients worsen weekly until reaching a state of akinetic mutism before death. The median survival is around 6 months and 90% of patients die within a year [1,2].
A small percentage of cases of sCJD have a longer survival [1-3]. This difference might be explained by the polymorphism at codon 129 of the prion protein gene and the type of prion strain [1-3].
We herein report the clinical, neuroimaging and pathological findings of a particularly long-duration case of sCJD.
Case Study
A 55-year-old man with cognitive complaints was referred to our department.
Ten months before he had left his job due to anxiety, depression and conflicts at work, but his family had noticed he was also experiencing cognitive problems. He did not remember what he had just done or where he had left things. He repeated the same questions and became lost even at home. His speech became less fluent. He did not know how to handle his mobile phone. He was not able to find the knob in the shower. He made mistakes when counting and dressing. His gait was awkward with frequent tripping and some falls.
Neurological examination showed frontal release signs (palmomental and grasp). The muscle tone was spastic, and the tendon reflexes were brisk with clonus. His gait was spastic without ataxia. Myoclonus was not noted. The MMSE score was 23/30 and a detailed neuropsychological examination detected failures in multiple cognitive domains (memory, executive function, praxis and visuospatial tasks). The patient showed anosognosia and difficulties in visual tracking. He scored positively on various items of NPI (anxiety, apathy, indifference, irritability and lability).
Laboratory findings were within normal ranges, including tests for syphilis, thyroid function, vitamins (A and E), ceruloplasmin, urine copper and immunological study. CSF revealed normal biochemistry and was negative for 14-3-3 protein. An initial electroencephalogram (EEG) showed little beta activity in the frontal areas without periodic discharges.
MRI (1.5 TESLA) showed hyperintense signals involving the dorsofrontal and parieto-occipital cortices and both medial thalamus which were particularly evident on DWI sequence. The motor and sensory primary cortices remained unaffected (Figure 1).
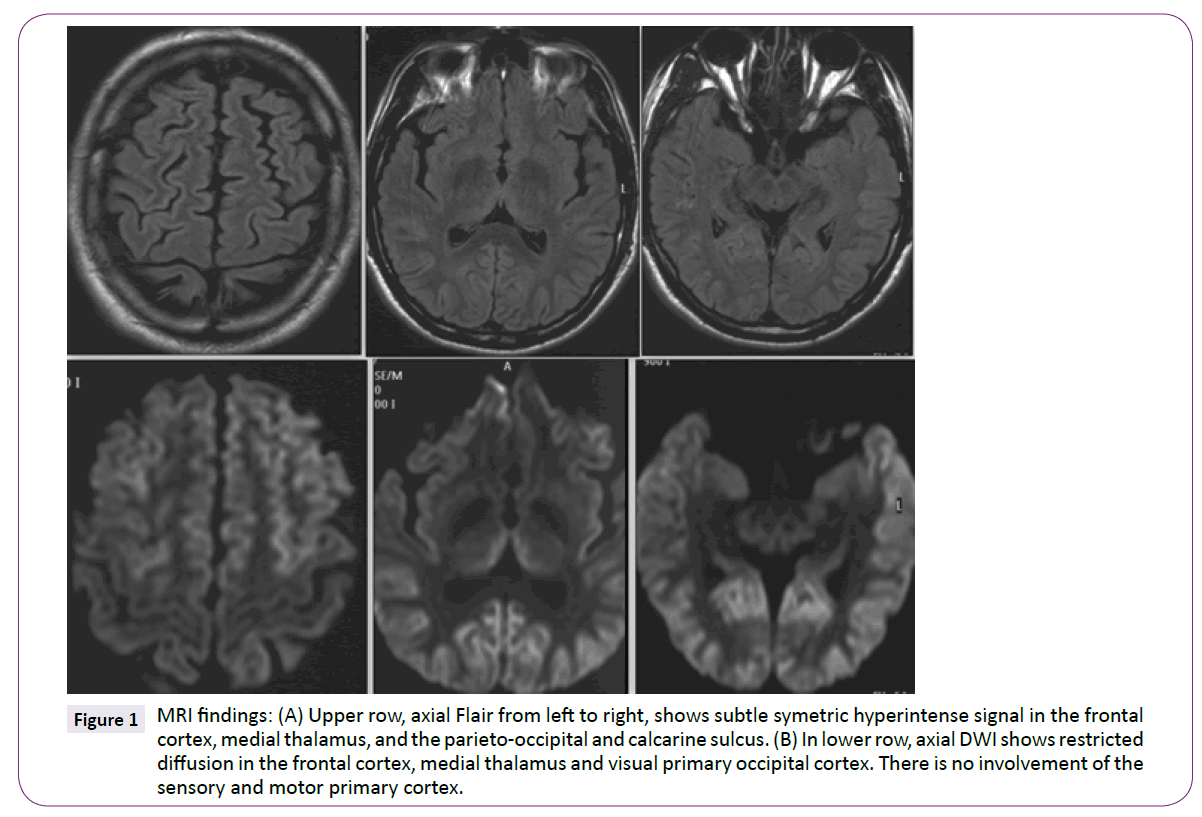
Figure 1: MRI findings: (A) Upper row, axial Flair from left to right, shows subtle symetric hyperintense signal in the frontal cortex, medial thalamus, and the parieto-occipital and calcarine sulcus. (B) In lower row, axial DWI shows restricted diffusion in the frontal cortex, medial thalamus and visual primary occipital cortex. There is no involvement of the sensory and motor primary cortex.
Genotyping of the prion protein gene did not reveal mutations. Codon 129 was heterozygous for methionine-valine (MV).
Seven months later the patient’s condition had worsened. He was suffering from agitation, visual hallucinations and false recognition delusions and was started on quetiapine up to 200 mg every day.
One year after the first evaluation the patient had double incontinence and his gait had worsened. On neurological examination, new signs had appeared such as supranuclear downgaze palsy, optic ataxia, facial hypomimia and rigidity. A new EEG showed generalised slowing without epileptiform discharges or periodic complexes.
Two years after the initial assessment the patient was no longer able to speak or move by himself. He had occasional irregular jerking in his limbs.
He died due to a respiratory infection in a state of akinetic mutism three years after the initial assessment and at least four years after the clinical onset.
Postmortem samples were taken from the cortical regions to confirm the diagnosis. Microscopic examination revealed typical diffuse spongiform changes with small vacuoles uniformly distributed all over the cortical layers, marked astrocytosis and severe neuronal loss. Characteristic Kuru-type plaques with dense PAS-positive centres were frequently identified (Figure 2A). An immunohistochemistry study showed an extensive accumulation of prion protein in a synaptic pattern with focal plaques (Figure 2B). Frozen tissue was not available for western blot analysis.
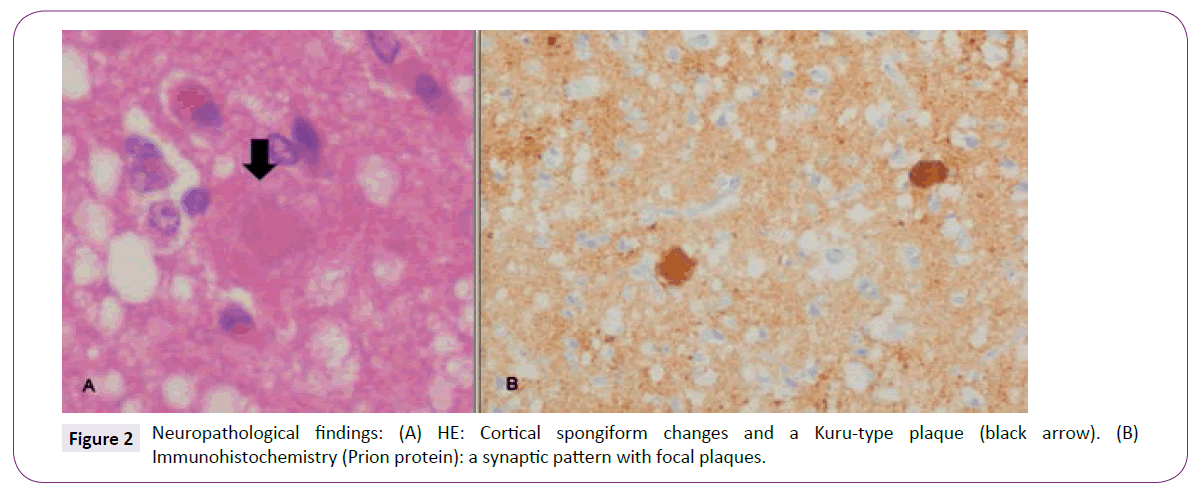
Figure 2: Neuropathological findings: (A) HE: Cortical spongiform changes and a Kuru-type plaque (black arrow). (B) Immunohistochemistry (Prion protein): a synaptic pattern with focal plaques.
Discussion
This patient was difficult to diagnose in life as sCJD because of the slow course of the disease (MMSE 23/30 a year after the disease onset, three years to reach a state of akinetic mutism and four years until death), the absence of periodic complexes on the EEG and the negativity for 14-3-3 protein on CSF. The diagnosis was based on the MRI findings until pathological samples could be obtained.
Within the spectrum of CJD there are some subtypes characterized by long-duration and negative EEG and 14-3-3 protein: variant Creutzfeldt-Jakob disease (vCJD) [4], some hereditary forms of CJD [5] and the more recently described “protease-sensitive prionopathy” [6]. The absence of pulvinar sign on MRI, the MV heterozygosis and the pathological study without amyloid florid plaques do not support vCJD. The prion protein gene analysis did not show mutations. The striking changes on DWI sequence and the neuropathological findings also discard “protease-sensitive prionopathy”.
Western-blot analysis to determine which type of prion strain the patient had was not available and this was the main limitation of our study. Nevertheless, the case shares a number of characteristics with previously described MV2 sCJD patients [1,3,7]: longduration (average duration of 17.1 months with a range from 5 to 72 months), negative EEG and 14-3-3 protein, medial thalamic hyperintensity on DWI sequence, Kuru-type plaques and synaptic patterns plus focal plaques on immunohistochemistry. However, some observations were atypical for MV2: absence of early ataxia, neuroimaging without involving striatum and uniform vacuolisation in all layers on neuropathology. The case may be classified as an atypical form of MV2 with severe cortical involvement, mimicking MM2-cortical-type sCJD, which has been described in other two cases up to now [8,9].
Sources of Financial Support
No funding received for this work
Conflicts of Interest
There are no conflicts.
7891
References
- Parchi P, Giese A, Capellari S (1999)Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46: 224-233.
- Parchi P, Saverioni D (2012) Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol50: 20-45.
- Krasnianski A, Schulz-Schaeffer WJ, Kallenberg K (2006) Clinical findings and diagnostic tests in the MV2 subtype of sporadic CJD. Brain 129: 2288-2296.
- Kaski D, Mead S, Hyare H (2009) Variant CJD in an individual heterozygous for PRNP codon 129. Lancet 374: 2128.
- Shimizu H, Yamada M, Matsubara N (2009) Creutzfeldt-Jakob disease with an M232R substitution: report of a patient showing slowly progressive disease with abundant plaque-like PrP deposits in the cerebellum. Neuropathology 29: 735-743.
- Gambetti P, Dong Z, Yuan J (2008) A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 63: 697-708.
- Krasnianski A, Kallenberg K, Collie DA (2008) MRI in the classical MM1 and the atypical MV2 subtypes of sporadic CJD: an inter-observer agreement study. Eur J Neurol15: 762-771.
- Samman I, Schulz-Schaeffer WJ, Wöhrle JC (1999) Clinical range and MRI in Creutzfeldt-Jakob disease with heterozygosity at codon 129 and prion protein type 2. J NeurolNeurosurg Psychiatry 67: 678-681.
- Ishihara K, Sugie M, Shiota J (2006)Severe cortical involvement in MV2 Creutzfeldt-Jakob disease: an autopsy case report. Neuropathology 26: 433-437.

