Keywords
Creutzfeldt-Jacob disease; Alien hand; Neurodegenerative disease
Introduction
Creutzfeldt–Jacob disease (CJD), a common spongiform encephalopathy, is a fatal neurodegenerative disease. There are four subtypes: familial, sporadic, iatrogenic, and variant CJD. In sporadic CJD, clinical symptoms of myoclonic jerks, rapidly progressive dementia, a typical electroencephalography (EEG) pattern, cranial magnetic resonance imaging (MRI) data and 14-3-3 positivity have important diagnostic value [1]. Besides the classical presentation, some neurodegenerative diseases presenting with rapidly progressive dementia such as Alzheimer’s disease, dementia with Lewy Bodies, frontotemporal dementia, meningoencephalitis, progressive supranuclear palsy, CADASIL, paraneoplastic encephalomyelitis, and corticobasal degeneration (CBD), can mimic CJD and make ia challenge in differential diagnosis [2].
We present a case of a 69-year-old female with asymmetrical dystonia and clumsiness of the left upper extremity, rapidly progressive cognitive decline, and other CBS symptoms. The patient was finally diagnosed with CJD based on EEG findings, radiological findings, and cerebrospinal fluid (CSF) data. This aim of this case report is to draw attention to the unusual clinical presentation of this CJD case.
Case Report
A 69-year-old, right-hand dominant female consulted our clinic with symptoms of clumsiness noticed by her family, in addition to weakness of her left arm and visual disturbance. She had presented to another outpatient clinic with symptoms of visual disturbance and dizziness 2 months earlier and all the clinical investigations were normal. Her family stated that when she tried to look directly at an object or reach for an object, she was unable to focus. She also encountered difficulties performing routine daily tasks such as using a knife, cooking and brushing her teeth. The patient had no specific medical history, and her physical examination was normal. She had left hemianopsia, left upper extremity dystonia, especially at adduction and flexion positions, and minimal rigidity. Her motor and sensory examinations were normal. The patient also had ideomotor apraxia, ideational apraxia, and very marked truncal ataxia, and after a few days she had developed left hemineglect. Based on these clinical symptoms, the patient was initially diagnosed with CBD.
The patient underwent detailed clinical examinations to formulate differential diagnoses. Diffusion weighted MRI (DWI) imaging depicted diffusion restriction at the right nucleus caudatus, anterior putamen, and bilateral parieto-occipital gyral areas, predominantly in the right hemisphere (Figures 1a and 1b). Routine laboratory investigations (a complete blood count, biochemistry, serology, etc.) were normal. Paraneoplastic markers and vasculitic biomarkers were all negative. In the examination of cerebrospinal fluid (CSF), protein and glucose levels were normal (glucose 82 mg/dl; protein 40 mg/dl), and no cells were detected. Given the rapid progressive clinical course and asymmetrical nucleus caudatus and cortical diffusion restrictions, CSF was sent to two independent laboratories for detection of the 14-3-3 protein biomarker. The results of both laboratories were positive. Serial EEG examinations were performed. The first EEG showed diffuse slow-wave baseline activity in both hemispheric regions. On subsequent EEGs, bilateral sharp and slow-wave paroxysmal activity (Figures 2a and 2b) was observed, in addition to irregular slow-wave activity. In light of the presence of diffuse myoclonia at follow up, the patient was started on levetiracetam therapy.
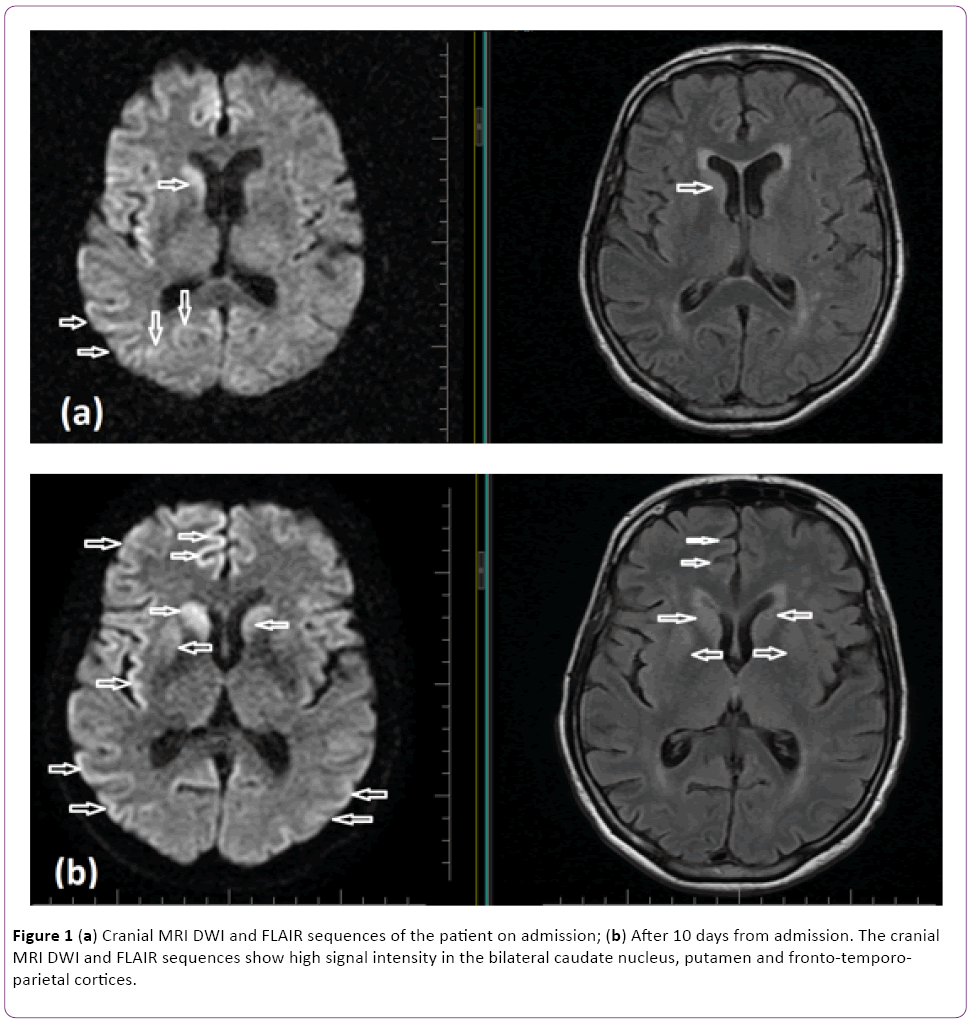
Figure 1: (a) Cranial MRI DWI and FLAIR sequences of the patient on admission; (b) After 10 days from admission. The cranial MRI DWI and FLAIR sequences show high signal intensity in the bilateral caudate nucleus, putamen and fronto-temporoparietal cortices.
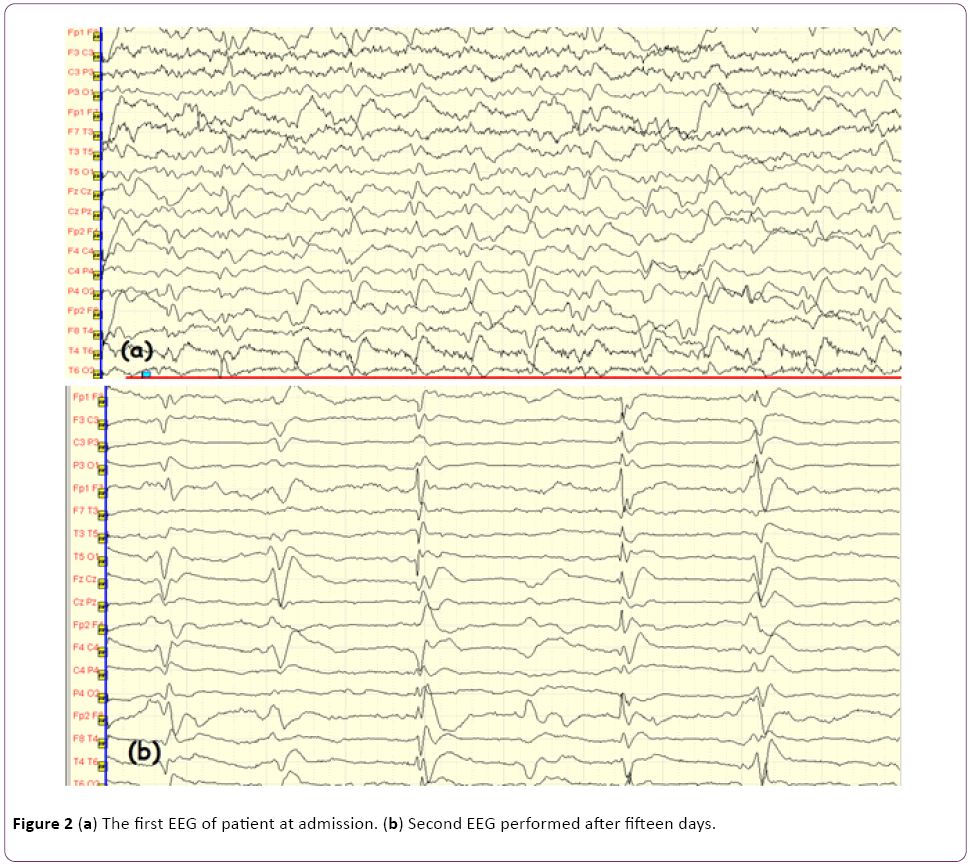
Figure 2: (a) The first EEG of patient at admission. (b) Second EEG performed after fifteen days.
Over the following weeks, in addition to the CBD symptoms, the patient developed dysphagia and speech difficulties. The patient was transferred to another hospital by her family after 2 months of clinical observation and died 4 months after the initial diagnosis.
Discussion
CJD is a rare fatal neurodegenerative disease and is the most frequent subtype of the spongiform encephalopathies. Sporadic CJD is the most common type (90% of cases) and mostly occurs between the ages of 50–70. It is characterized by rapidly progressive multifocal dementia, with myoclonia [3]. Sporadic CJD is also associated with personality changes, cerebellar system symptoms other than dementia, and ocular system symptoms. Vocal or tactile myoclonia accompanies the symptoms in the majority of patients. Pyramidal, extrapyramidal system disorders, epileptic seizures, and akinetic mutism can also develop at later stages of the disease [4,5].
CJD is caused by prion proteins. Basic characteristic of all prion diseases is the proliferation of prion proteins (PrP) from a normal isoform (PrPc protein) to an abnormal isoform (PrPs protein). The latter proteins then accumulate in the brain and cause infection [6].
EEG is one of the most important investigations, with 95% diagnostic value in CJD. Baseline activity slowing, in addition to diffuse and morphologically triphasic periodic sharp-wave, provides important evidence [7]. In the present case, serial EEG recordings were performed. The first EEG revealed abnormal baseline activity and dismorphic sharp-wave changes. Fifteen days later, a control EEG showed typical 1 Hz/sec periodic sharp-wave discharges.
Cranial MRI is also very valuable in the diagnosis of CJD. The signal hyperintensity in the cortical and deep gray matter of the brain in T2A and FLAIR sequences and diffusion restrictions in DWI are typical of the sporadic form of the disease. The nucleus caudatus and putamen, insula, cingulate, and superior frontal gyri are usually affected [8]. In variant CJD, the signal hyperintensity at the pulvinar thalamus is pathognomonic and is known as the “pulvinar sign” or “hockey-stick sign”. DWI screening is accepted as the most sensitive diagnostic MRI sequence for CJD [9].
In the current case, on the first cranial MRI, hyperintensity was detected in the right nucleus caudatus, bilateral frontal cortical and parieto-occipital on the diffusion-weighted and FLAIR sequences. As the disease progressed, the bilateral nucleus caudatus was also affected, and the hyperintensities of the frontal cortical and parieto-occipital regions were more significant (Figures 1a and 1b).
The detection of the 14-3-3 protein in the CSF supported the diagnosis of CJD. The diagnostic specificity and sensitivity of the positivity of this protein for CJD were both reported to be 92% [10].
CBD is generally a slow, progressive disease. However, occasionally, it may start abruptly and progress rapidly therefore can easily be confused with CJD. The cerebral cortex and basal ganglia are affected in both diseases so the clinical symptoms can be similar [11]. Limb clumsiness is the most common presentation in CBD (67%) while isolated sensory symptoms occurred in only 8%. On the contrary, limb sensory disturbance was the most common initial complaint in CJD (50%) followed by limb clumsiness (35%) [11]. In CBD, myoclonus and alien hand occurred after one year from disease onset but much more earlier in CJD patients [11].
The clinical presentation of the disease begins with asymmetrical extrapyramidal symptoms, such as Parkinsonism, extremity dystonia, postural instability, and symptoms of cortical involvement. The latter includes various symptoms, such as apraxia, cortical desensitization, and alien hand syndrome. Postural or action tremor can also be seen. Besides these commonly encountered symptoms, less common symptoms, such as reflex myoclonus, atetosis, orolingual dyskinesia, frontal lobe primitive reflexes, eye movement disorders, dysarthria, speech apraxia, dementia, and hyperreflexia may also accompany the clinical picture [12].
In Lee W. and his friend’s study of 20 patients diagnosed as CBD-CJD, the most common initial symptoms were apraxia (70%) and alien hand (85%). The most common movement disorders were myoclonia (80%) and dystonia (45%). The duration from disease onset to death was averagely 5 months. The 53% of the patients had typical EEG pattern. 17 patients were performed cranial MRI and 7 of them restricted diffusion at cerebral cortices and basal ganglia [13].
In the present case, the initial diagnosis was CBD because of the presence of asymmetrical dystonic posture and apraxia of the left arm and hand, cortical desensitization deficits at the left side of the body, asymmetrical rigidity, and Parkinsonism symptoms. However, at the 2-week follow up, myoclonia, dysphagia, and speech disorders were observed, and these symptoms progressed rapidly. As a result, the final diagnosis was CJD. The patient’s condition continued to deteoriate, and she died 4 months after the initial diagnosis.
Conclusion
In summary, CJD is a rapidly progressive degenerative disease with a broad spectrum of clinical symptoms. The characteristic EEG and cranial MRI data and the 14-3-3 protein positivity in CSF are of great importance whenever the differential diagnosis cannot be based on clinical symptoms. CJD can mimic all other neurodegenerative diseases and it progresses rapidly therefore to achieve correct diagnosis is vital to adjust the treatment plan and follow up.
17975
References
- Inzelberg R, Nisipeanu P, Blumen SC, Carasso RL (2000)Alien hand sign in Creutzfeldt–Jacob disease. NeurolNeurosurgPsychiatry 68: 100–126.
- Kojima G, Tatsuno BK, Inaba M, Velligas S, Masaki K, et al. (2013) Creutzfeldt-Jacob disease: A case report and differential diagnoses.Hawaii J Med Public Health 72: 136-139.
- Johnson RT, Gibbs CJ Jr (1998) Creutzfeldt–Jacob disease a related transmissible spongiform encephalopathies.N Engl J Med 339: 1994–2000.
- Wadsworth JDF, Collinge J (2011) Molecular pathology of human prion disease. ActaNeuropathol 121: 69.
- Tschamba HJ, Zerr I, Urbach H (2007) Radiological assessment of Creutzfeldt–Jacob disease. EurRadiol 17: 1200–1211.
- Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J (1996) Diagnostic criteria for sporadic Creutzfeldt–Jacob disease. Arch Neurol 53: 913–920.
- Wieser HG, Schindler K, Zumsteg D (2006) EEG in Creutzfeldt–Jacob disease. ClinNeurophysiol 117: 935–951.
- Kallenberg K, Schulz-Schaeffer WJ, Jastrow U, Poser S, Meissner B, et al. (2006) Creutzfeldt–Jacob disease: Comparative analysis of MR imaging sequences ANJR 27: 1459–1462.
- Collie DA, Summers DM, Sellar RJ, Ironside JW, Cooper S, et al. (2003) Diagnosing variant Creutzfeldt–Jacob disease with the pulvinar sign: MR imaging findings in 86 neuropathologically confirmed cases. AJNR Am J Neuroradiol 24: 1560–1569.
- Zerr I, Pocchiari M, Collins S, Brandel JP, De Pedro-Cuesta J, et al. (2000) Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt–Jacob disease. Neurology 55: 811–815.
- ValverdeAH, Costa C, Timoteo A, Ginestal R, Pimentel J (2011) Rapidly progressive corticobasal degeneration syndrome. Case Rep Neurol 3: 185-190.
- Riley DE, Lang AE (1996) Movement disorders. In: Bradley WG,(ed) Neurology in clinical practice. (2ndedn). Boston:Butterworth-Heinemann 1996: 1889–1951.
- Lee W, Simpson M, Ling H, McLean C, Collins S, et al. (2013) Characterizing the uncommon corticobasal syndrome presentation of sporadic Creutzfeldt-Jacob disease. Parkinsonism RelatDisord 19: 81-85.

