Keywords
Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs); β-lactamases inhibitors; Antibacterial resistance; Drug discovery; Enzyme inhibition
Introduction
The alarming rate of β-lactamase-mediated resistance to β-lactam antibiotics has emerged as a significant clinical threat to almost all of these life-saving drugs. The rapid resistance adaptation of bacterial strains to these antibiotic is the result of selection pressure induced by overuse and abuse of β-lactam antibiotics [1,2]. Bacteria have developed resistance to antibiotics in two major ways; (i) the production of β-lactamases, which is often time accompanied by a decrease in outer membrane permeability and (ii) the production of low-affinity drug resistant penicillin-binding proteins (PBPs) [3,4]. As a result, most classic β-lactam antibiotics are becoming progressively less effective as antimicrobial agents. It has been shown that a successful strategy for combating β-lactamase induced resistance is the increase of agents designed to bind competitively at the active site of β-lactamases thus inhibiting the enzyme from hydrolyzing β-lactams [5].
Aside from the regular β-lactam based β-lactamase inhibitors (clavulanic acid, sulbactam, and tazobactam) which have been used successfully in combination with penicillins and cephalosporins for last three decades, scientists have focused their attention on the synthesis and isolation of effective non-β-lactam inhibitors. These antiresistance stewardship agents are being administered as antibiotic adjuvants to potentiate the effects of current antimicrobials in bacteria where they are no longer effective and serve as suicide inhibitors to assist the regular β-lactams to gain access to their target active sites without being hydrolyzed [6]. The non-β-lactam inhibitors that work in conjunction with β-lactams are classified into three main groups: transition state analogs, substrate analogs, and non-covalent inhibitors [7]. Avibactam (NXL104; AstraZeneca) and PRX-7009 are examples of recently developed, highly effective non-β-lactam based β-lactamase inhibitors (Figure 1) [8,9]. To date, avibactam is the first non-β-lactam β-lactamase inhibitor which was approved by the FDA in February 2015 in combination with Ceftazidime [10]. It is noteworthy to recall that although the hundreds of compounds reported having the ability to inhibit several kinds of β-lactamases, coevolution never stops and bacteria have a strong capacity to find a way to survive this particular adjuvant strategy leading to the emergence of resistance. Therefore, the necessity to constantly attempt to find new classes of efficient non-β-lactam inhibitors remains vital for antimicrobial stewardship programs to take a continuing lead role in their appropriate drug discovery and development.
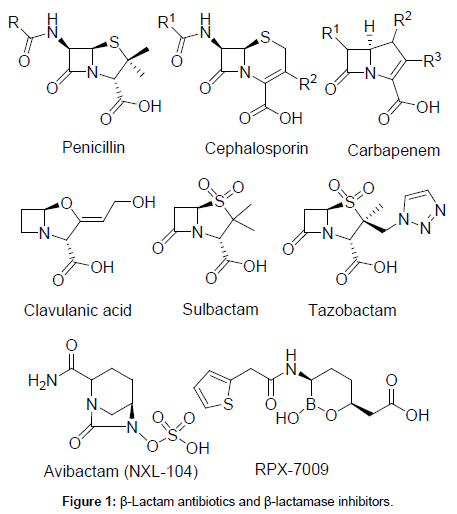
Figure 1: β-Lactam antibiotics and β-lactamase inhibitors.
Due to limited front-line antibacterial chemotherapeutic agents and emergence of resistance to the last-resort antibiotic class, carbapenems and colistin, there has been a renewed interest in the revival of agents that exert their antibacterial effects by binding selectively to specific sequences of duplex DNA [11]. Pyrrolo[2,1-c][1,4]benzodiazepine (PBD) class of compounds has sparked a great interest to evaluate as potential DNA-interactive antibacterial agents [12,13]. PBDs are a family of naturally occurring antitumor antibiotics that display their cytotoxic activity by recognizing and binding covalently to the C-2 amino group of guanine residues within the minor groove of DNA [14,15]. PBD/DNA interaction assumed to occur in two stages, with initially by noncovalent recognition of its binding pocket within the wall and floor of the minor groove, followed by nucleophilic attack of guanine C2-NH2 on the N10-C11 imino group [16]. PBD monomers span three DNA base pairs with a thermodynamic preference for Pu-GPu (where Pu=purine; G=guanine) sequences and block transcription by inhibiting RNA polymerase activity in a sequence-specific manner [17]. Some of the naturally occurring PBDs such as tomaymycin, anthramycin, and DC-81 display their potent antibacterial activity against human pathogens through a capacity to bind to DNA (Figure 2) [18-20]. However, a high degree of cytotoxicity has made them less attractive as an antibacterial agent. Sibiromycin and anthramycin have been shown cardiotoxicity and tissue necrosis in animal models due to the possible free radical formation by the C9-OH group and production of o-quinone imine species by oxidation [21,22].
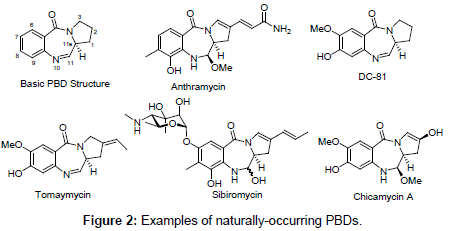
Figure 2: Examples of naturally-occurring PBDs.
Previously reported capability of the parent PBD molecule and its synthetic analogs to act on bacterial cells’ DNA unveil their ability to cross the outer membrane of microorganisms thus making them accessible to the target which is the cytoplasmic membrane. This feature makes them promising candidates for new non-β-lactam β-lactamase inhibitors [23-27]. The long-term goal of my research group is to discover novel non-β-lactam inhibitors of β-lactamases via design, synthesis, and evaluation of new chemotypes, in order to find new ways of combating β-lactamase induced multidrug antibacterial resistance. The ideal inhibitors should possess chemical features such as enhanced permeability through the cell membrane, affinity for the active site of β-lactamase, formation of intermediates that traps the enzyme, displacement of critical water molecules and prolonged time to enzyme recovery [28].
Despite a high level of interest on PBDs as lead candidates for the preparation of antimicrobial compounds due to their cancerostatic and anti-infective properties, there is not a single report related to determination of their efficacy as serine β-lactamase inhibitors. Thus, a structure-based rational design is employed here to guide the creation of novel PBD-based β-lactamases inhibitors. A closer inspection of marketed serine β-lactamase inhibitors, as well as their evolutionary relatives, penicillins and carbapenems, revealed a common 7-ring member 1,4-diazepine-5-one (I) scaffold after intramolecular C-N bond cleavage on bicyclic molecules (Figures 1 and 3). This similarity makes it capable of possibly interacting with the enzyme active sites of transpeptidases and β-lactamases which are involved in cell wall synthesis and antibiotic bacterial resistance, respectively.
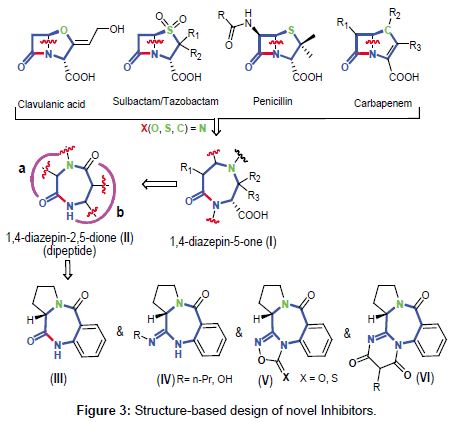
Figure 3: Structure-based design of novel Inhibitors.
In an effort to discover novel non-β-lactam inhibitors that interact with key active-site residues, we propose that construction of a cyclic dipeptide/dilactam chemotype [1,4-diazepin-2,5-dione dilactam (II)], could likely serve as a core structure to adopt a serine-γ-OH nucleophilic attack to its electrophilic site within the active site of β-lactamases. Dilactam II is composed of two modules: a (α-amino acids) and b (anthranilic acid). We will start our project with the synthesis and evaluation of pyrrolo[2,1-c][1,4]benzodiazepine (PBD) III, followed by electronically activated tetracyclic PBD analogs (IVVI). In particular, our goal is to establish the structural requirement to fulfill the optimal in vitro inhibitory activity, whereas sustaining a reduced toxicity, unlike other present PBD products. To date, there is no report of β-lactamases inhibitors from the PBD class of compounds.
Materials and Methods
A Perkin-Elmer Spectrum Series FTIR spectrometer with an attenuated total reflectance adapter was used for Infra-Red studies. An Agilent Technologies Cary 8454 UV/Vis. spectrophotometer with a PCB-1500 water Peltier circulating system by Agilent Technologies and quartz 1000 μL cuvettes with a path length of 1 cm were used for UV/Vis. absorbance and kinetics studies. Melting Points was measured using a Thermo Scientific Electrothermal Digital Melting Point Apparatus IA9100 series and molecular weight determination was done using a Shimadzu GC-MS-QP 2010 Plus. The 1H and 13C NMR spectra were recorded in DMSO-d6 and CHCl3-d on a JEOL Eclipse 400 MHz NMR Spectrometer operating at 400 MHz for 1H and 100 MHz for 13C NMR. Chemical shift (δ) values are expressed in parts per million (ppm) and are referenced to the residual solvent signals of DMSO-d6 and CHCl3-d at δH/δC 2.50/39.5 and 7.25/76.8, 77.1, 77.4, respectively. Optical rotations were measured with a Roudolph Research Analitical AUTOPOL® III digital polarimeter. TLC analysis was carried out on precoated silica gel G254 aluminum plates. Nitrocefin (NCF) as β-lactamase substrate was purchased from BioVision Inc. TEM-1 β-lactamase was purchased from Invitrogen (Life Technologies). P99 β-lactamase from Enterobacter cloacae was purchased from Sigma Aldrich. All chemical reagents and solvents were purchased from Acros Organic, Alfa Aesar Chemical Company, and Sigma-Aldrich Chemical Company and used without further purification. The positive control, clavulanic acid (CLA) was purchased from Sigma-Aldrich Chemical Company.
Experimental section
(S)-1,2,3,11a-Tetrahydro-5H-benzo[e]pyrrolo[1,2-c][1,4] diazepine-5,11(10H)-dione (1): In a 250 mL one-neck round bottom flask, a suspension of isatoic anhydride (20.0 g, 122.7 mmol) and L-proline (14.12 g, 122.6 mmol) in DMF (60 mL) was heated to 155°C for 5 hours. The solvent was removed in vacuo, and the residue was taken up in cold water. The precipitate was collected and dried to give the dilactam. The resultant solid was purified by recrystallization through slow evaporation from acetone/DMF (10:1) to afford pure colorless crystals in very good yield.
Yield: 24.78 g (93.6%). m.p.: 223 - 225°C. 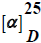 =+512º (c=0.5, CH3OH).
=+512º (c=0.5, CH3OH).
1H-NMR (400 MHz, DMSO-d6): δ = 1.76-2.00 (m, 4H), 3.42-3.48 (m, 1H), 3.56-3.61 (m, 1H), 4.10 (d, 1H), 7.11-7.13 (dd, 1H), 7.20-7.24 (m, 1H), 7.49-7.51 (m, 1H), 7.77-7.79 (dd, 1H), 10.51 (s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 23.6, 26.3, 40.4, 56.7, 121.8, 124.4, 127.1, 130.8, 132.6, 136.9, 165.0 (CO), 171.3 (CO). IR (KBr): ν̃ (cm-1) = 3222 (NH), 3206, 2955, 2918, 2850, 1691 (CO), 1680 (CO), 1621, 1551, 1536, 1479, 1443, 1412, 1385, 1285, 1259, 1179, 759, 701, 615. UV λmax (MeOH): 198, 274 nm. GC-MS (70 eV) m/z (%): 216 (10) [M+], 119 (14), 92 (20), 70 (100), 64 (10).
(S)-11-Thioxo-1,2,3,10,11,11a-hexahydro-5H-benzo[e] pyrrolo[1,2-c][1,4]diazepin-5-one (2): In a 250 mL one-neck round bottom flask, a mixture of dilactam (2.15 g, 10.0 mmol) and Lawesson’s reagent (4.04 g, 10.0 mmol) in THF (100 mL) was stirred for 24 hours at room temperature. Evaporation of the solvent in vacuo gave a yellow solid residue which was purified by dissolving in toluene and filtered off by gravity. The solid was further washed with cold toluene to obtain pure yellow product. Further purification was carried out by crystallization through slow evaporation from acetone/DMF (20:1) to afford pure yellowish crystals in very good yield.
Yield: 2.03 g (88.0%). m.p.: 272-274 °C. =+762º (c=0.5, CHCl3).
1H-NMR (400 MHz, DMSO-d6): δ = 1.98-1.83 (m, 1H), 1.98- 2.15(m, 2H), 2.88 (d, J=5.9 Hz, 1H), 3.42-3.48 (m, 3H), 3.56-3.61 (m, 1H), 4.28 (d, J=6.2 Hz, 1H), 7.27-7.29 (dd, J=8.1 Hz, 1H), 7.33-7.37 (m, 1H), 7.55-7.60 (ddd, J=7.7, 1.2 Hz, 1H), 7.82-7.84 (dd, J=7.7, 1.5 Hz, 1H), 8.13 (s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6): δ = 23.2, 29.5, 47.4, 60.3, 122.3, 126.2, 128.3, 130.8, 132.7, 137.0, 164.7 (CO), 202.5 (CS). IR (KBr): ν̃ (cm-1) = 3125 (N-H), 3094, 3063, 3024, 2974, 1620 (C=O), 1579, 1523, 1478, 1452, 1418, 1381, 1272, 1193, 1166, 1145, 1103, 1069, 1055, 887, 833, 817, 786, 755, 695, 664, 625. UV λmax (MeOH): 194, 274 nm. GC-MS (70 eV) m/z (%): 232 (7) [M+], 108 (6), 70 (100), 68 (6).
(S)-11-(Propylamino)-1,2,3,11a-tetrahydro-5H-benzo[e] pyrrolo[1,2-c][1,4]diazepin-5-one (3): To a stirred suspension of monothiolactam (5.78 g, 25.0 mmol) and propylamine (20 mL) was added HgCl2 (7.14 g, 26.25 mmol) at 60°C. The mixture was stirred for 1 hour at this temperature. After cooling to room temperature, the mixture was filtered through a plug of celite and washed with CH2Cl2. The filtrate was washed with sat. Na2S2O3(aq) and dried over MgSO4, filtered, and the solvent and excess amine were evaporated under reduced pressure. The resultant solid was purified by recrystallization from nitromethane to afford pure colorless crystals in very good yield.
Yield: 5.64 g (88%). m.p.: 159-161°C. =+1106º (c=0.5, CHCl3)
1H-NMR (400 MHz, CDCl3-d6): δ = 0.96-0.99 (t, J=7.5 Hz, 3H), 1.64-1.69 (m, 2H), 1.99-2.12 (m, 2H), 2.20-2.25 (m, 2H), 3.37-3.38 (d, J=4.8 Hz, 2H), 3.50-3.60 (m, 1H), 3.84-3.88 (m, 1H), 4.01-4.03 (t, J=4.9 Hz, 1H), 4.68 (s, 1H), 7.03-7.05 (m, 1H), 7.07-7.11 (m, 1H), 7.36-7.38 (ddd, J=8.6, 6.8, 1.3 Hz,1H), 7.91-7.93 (dd, J=7.7, 1.5 Hz, 1H). 13C-NMR (400 MHz, CDCl3): δ = 11.7, 22.3, 23.9, 26.8, 43.3, 46.5, 54.4, 122.2, 126.5, 126.9, 130.1, 131.6, 146.7, 156.2 (CN), 166.8 (CO). IR (KBr): ν̃ (cm-1) = 3851, 3798, 3745, 3356 (N-H), 3319 (N-H), 3287, 3061, 2941, 2880, 2815, 2359, 2328, 1826, 1791, 1731, 1605 (C=O), 1554, 1531, 1506, 1456, 1406, 1383, 1336, 1256, 1215, 1150, 1096, 1067, 1035, 991, 918, 835, 761, 703, 635. UV λmax (MeOH): 198, 273 nm. GC-MS (70 eV) m/z (%): 257 (21) [M+], 146 (23), 119 (23), 90 (21), 70 (100).
(14aS)-3-Phenyl-1-propyl-1,12,13,14,14a,14b-hexahydro- 2H,10H-benzo[e]pyrimido[2,1-c]pyrrolo[1,2 c][1,4]diazepine- 2,4,10(3H)-trione (4): A mixture of 3 (0.257 g, 1.0 mmol) and bis(2,4,6-trichlorophenyl)-2-phenylmalonate (0.539 g, 1.0 mmol,) was heated at 190°C for 10 minutes in a Zincke apparatus under high vacuum. The residue was treated with diethyl ether to give a dark brown precipitate which was collected by filtration and washed with diethyl ether. Recrystallization was done from DMF/Water to yield pale yellow single crystals.
Yield: 300 mg (75%) m.p.: 230 - 233 °C. =0º (c=0.5, CHCl3)
1H-NMR (400 MHz, CDCl3): δ = 0.77-0.81 (t, J=7.3 Hz, 3H), 1.31- 1.39 (m, 2H), 2.04-2.17 (m, 1H), 2.21-2.28 (m, 1H), 2.62-2.71 (m, 1H), 2.77-2.82 (dd, J=14.6, 6.2 Hz, 1H), 2.96-3.03 (m, 1H), 3.96-4.11 (m, 2H), 4.17-4.25 (m, 1H), 4.71 (s, 1H), 7.28-7.30 (m, 2H), 7.36-7.45 (m, 5H), 7.54-7.59 (td, J=7.8, 1.6 Hz, 1H), 8.02-8.04 (m, 1H). 13C-NMR (100 MHz, CDCl3): δ = 11.2, 20.6, 21.5, 29.6, 48.4, 49.5, 59.1, 118.0, 125.5, 127.5, 128.2, 128.4, 130.8, 131.3, 132.3, 133.3 (CO), 139.8 (CO), 167 (CO). IR (KBr): ν̃ (cm-1) = 2957, 2918, 2860, 2355, 1958, 1728, 1683 (C=O), 1633 (C=O), 1576, 1487, 1453, 1352, 1297, 1252, 1221, 1185, 1150, 899, 800, 753, 719, 698, 665, 631. UV λmax (AcO): 194, 274 nm. GC-MS (70 eV) m/z (%): 401 (66) [M+], 215 (11), 187 (29), 118 (100), 90 (46).
(S)-11-(Hydroxyamino)-1,2,3,11a-tetrahydro-5H-benzo[e] pyrrolo[1,2-c][1,4]diazepin-5-one (5): Hydroxylamine hydrochloride (800 mg, 11.51 mmol) and potassium carbonate (6.0 g, 43.5 mmol) was added to a solution of monothiolactam (1.74 g, 7.5 mmol) in absolute ethanol (40 mL) and stirred for 48 hours at room temperature. The initial yellow mixture decolorized and H2S was released. The mixture was taken up in dichloromethane (120 mL) and washed with water (80 mL). The organic layer was further washed with brine and dried over Na2SO4. The solvent was removed in vacuo. The crude residue was dissolved in a mixture of diethyl ether/hexane (1:1), filtered and further washed in 20 mL of the solvent mixture to obtain 1.63 g of the pure solid. Recrystallization was done from nitromethane to produce offwhite crystals.
Yield: 1.63 g (94%). m.p.: 150 - 153 °C. =+488º (c=0.5, CDCl3)
1H-NMR (400 MHz, DMSO-d6): δ = 1.86-2.03 (m, 3H), 2.56-2.60 (m, 1H), 3.47-3.61 (m, 4H), 4.31-4.33 (m, 1H), 7.03-7.06 (t, J=7.5 Hz, 1H), 7.25-7.27 (dd, J=16.8, 7.7 Hz, 1H), 7.37-7.41 (t, J=7.7 Hz, 1H), 7.66-7.68 (d, J=7.7 Hz, 1H), 8.75 (s, 1H, N-H), 10.08 (s, 1H, OH). 13C-NMR (100 MHz, CDCl3-d6): δ = 23.4, 25.9, 47.4, 54.4, 120.5, 123.5, 125.8, 131.6, 132.6, 136.9, 151.2, 166.2 (CO). IR (KBr): ν̃ (cm-1) = 3788, 3716, 3281, 2965, 2911, 2878, 2806, 2351, 1724, 1689, 1658, 1612, 1573, 1552, 1530, 1480, 1453, 1425, 1396, 1273, 1227, 1201, 1162, 1108, 1040, 997, 957, 933, 884, 847, 807, 787, 756, 701, 663. UV λmax (MeOH): 198, 313 nm. GC-MS (70 eV) m/z (%): 231 (20) [M+], 144 (37), 90 (35), 70 (100).
(S)-11,12,13,13a-Tetrahydro-3H,9H-benzo[e][1,2,4] oxadiazolo[3,4-c]pyrrolo[1,2-c][1,4]diazepine-3,9-dione (6): Under nitrogen atmosphere, a solution of 5 (231 mg, 1.0 mmol) in anhydrous dioxane (8 mL) was added to 1,1-carbonyl diimidazole (CDI) (487 mg, 3.3 mmol). The reaction mixture was refluxed for 12 hours and the solvent removed in vacuo. The residue was taken up in dichloromethane and washed three times with water. The organic layer was dried over Na2SO4 and the solvent was removed under reduced pressure. The crude residue was purified by flash column chromatography to obtain a white solid which was recrystallized from diisopropyl ether /ethyl acetate (1:1) to get crystals used for x-ray crystallography.
Yield: 226.16 mg (88%). m.p.: 180 - 182°C. =+142º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3-d6): δ = 1.58 (d, J=12.1 Hz, 2H), 2.10- 2.23 (m, 2H), 2.30-2.39 (m, 1H), 2.82-2.87 (tt, J=9.9, 3.4 Hz, 1H), 3.67- 3.74 (m, 1H), 3.90-3.95 (m, 1H), 4.58-4.61 (dd, J=8.4, 2.9 Hz, 1H), 7.49- 7.53 (m, 1H), 7.64-7.68 (td, J=7.8, 1.6 Hz, 1H), 7.83-7.85 (d, J=7.3 Hz, 1H), 8.02-8.04 (dd, J=7.9, 1.6 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ = 23.4, 25.6, 47.9, 51.2, 122.4, 128.6, 128.6, 128.7, 132.8, 156.3 (CN), 158.0 (CO), 164.2 (CO). IR (KBr): ν̃ (cm-1) = 3623, 3335, 3044, 2956, 2918, 2875, 2851, 2381, 2349, 2296, 2199, 2105, 1981, 1838, 1787, 1728, 1710, 1690, 1657, 1640, 1599, 1551, 1468, 1451, 1410, 1301, 1267, 1167, 1081, 1025, 990, 761, 702, 662, 608. UV λmax (MeOH): 198, 274 nm. GC-MS (70 eV) m/z (%): 257 (22) [M+], 144 (100),116 (38), 90 (33), 44 (33), 41 (24).
(S)-3-Thioxo-11,12,13,13a-tetrahydro-3H,9H-benzo[e][1,2,4] oxadiazolo[3,4-c]pyrrolo[1,2-c][1,4]diazepin-9-one (7): A solution of 5 (231 mg, 1 mmol) in anhydrous dioxane (8 mL) was added to 1,1-thionyl diimidazole (TDI) (486 mg, 3.3 mmol) under nitrogen atmosphere at room temperature. The reaction mixture was refluxed for 12 hours and the solvent removed in vacuo. The residue was taken up in dichloromethane and washed three times with water. The organic layer was dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified by flash column chromatography to obtain a pale yellow solid followed, by cystallization from diisopropyl ether/ ethyl acetate (1:1) to afford pale yellow crystals.
Yield: 245.7 mg (90%). m.p.: 216 - 218°C. =+34º (c=0.5, CDCl3).
1H-NMR (400 MHz, CDCl3): δ = 1.17-1.27 (m, 1H), 2.06-2.29 (m, 2H), 2.32-2.47 (m, 1H), 2.84-2.90 (m, 1H), 2.88 (tt, J=9.9, 3.3 Hz, 1H), 3.65-3.72 (m, 1H), 3.90-3.95 (m, 1H), 4.57-4.60 (dd, J = 8.6, 2.7 Hz, 1H), 7.55-7.59 (td, J=7.6, 0.9 Hz, 1H), 7.67-7.72 (m, 1H), 8.01-8.04 (dd, J=8.1, 1.5 Hz, 1H), 8.27-8.29 (dd, J=8.2, 0.9 Hz, 1H). 13C-NMR (100 MHz, CDCl3): δ = 23.5, 26.5, 47.6, 51.0, 124.7, 129.8, 131.8, 132.2. IR (KBr): ν̃ (cm-1) = 3788, 3716, 3281, 2965, 2911, 2878, 2806, 2351, 1724, 1689, 1658, 1612, 1573, 1552, 1530, 1480, 1453, 1425, 1396, 1273, 1227, 1201, 1162, 1108, 1040, 997, 957, 933, 884, 847, 807, 787, 756, 701, 663. UV λmax (MeOH): 198, 276 nm. GC-MS (70 eV) m/z (%): 273 (70) [M+], 146 (42), 102 (75), 90 (65), 69 (44), 43 (100).
N-phenylethane thioamide (8): In a 250 mL of one-neck round bottom flask, a mixture of acetanilide (6.75 g, 50.0 mmol) and Lawesson’s reagent (10.1 g, 25.0 mmol) in DCM (100 mL) was stirred for 5 hours at room temperature. Evaporation of solvent in vacuo gave a yellow solid residue which was purified by column chromatography using DCM.
Yield: 7.02 g (93%). m.p.: 74 - 76°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ = 2.16 (s, 3H), 7.08-7.11 (t, 1H), 7.28-7.32 (t, 2H), 7.48-7.50 (d, J=7.7 Hz, 1H), 13C-NMR (100 MHz, CDCl3): δ = 24.7, 120, 124.4, 129.1, 139, 169.6 (CO), IR (KBr): ν̃ (cm- 1) = 3184, 3164, 3002, 2957, 2920, 2359, 1595, 1533, 1495, UV λmax (MeOH): 198, 277 nm. GC-MS (70 eV) m/z (%): 151 (43) [M+], 110 (54), 93 (100), 77 (100), 59 (76).
(E)-N’-Phenyl-N-propylacetimidamide (9): To a stirred suspension of 8 (755 mg, 5 mmol) and propylamine (20 mL, 59.11 mmol) was added HgCl2 (1.42 g, 5.25 mmol) at 60°C. The mixture was stirred for 1 hour at this temperature. After cooling to room temperature, the mixture was filtered off through a plug of celite and eluted with CH2Cl2. The filtrate was washed with sat. Na2S2O3(aq) and dried over MgSO4, filtered, and the solvent and excess amine were evaporated under reduced pressure. The resultant solid was purified by fractional distillation to yield a brownish liquid which solidifies on refrigeration.
Yield: 619.5 mg (70%). m.p.: 180-182°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ = 0.96-1.00 (m, 3H), 1.19 (s, 1H), 1.57-1.66 (m, 2H), 1.77 (s, 3H), 3.28-3.32 (t, J=4.4 Hz, 2H), 6.75-6.77 (m, 2H), 6.94-6.97 (d, J=6.6 Hz,1H), 7.21-7.29 (m, 2H). 13C-NMR (100 MHz, CDCl3): δ = 14.3, 21.5, 30.4, 60.5, 122.6, 128.8, 131, 171.3, 207.2. IR (KBr): ν̃ (cm-1) = 3419, 3282 (NH), 2961, 2929, 2872, 2362, 1626, 1592, 1542, 1487, 1382, 1261, 1223, 1168, 1070, 900, 800, 743, 699. UV λmax (MeOH): 194, 274 nm. GC-MS (70 eV) m/z (%): 176 (19) [M+], 133 (16), 118 (73), 93 (96), 77 (100), 59 (30), 42 (56).
2-Methyl-1,5-diphenyl-3-propyldihydropyrimidine- 4,6(1H,5H)-dione (10): A mixture of 9 (176 mg, 1.0 mmol) and bis(2,4,6-trichlorophenyl)-2-phenylmalonate (539 mg 1.0 mmol) was heated at 100°C for 10 minutes in a Zincke apparatus under high vacuum. The residue was treated with diethyl ether to give a dark brown precipitate which was collected by filtration and washed with diethyl ether. Recrystallization was done from 95% ethanol/ 2-propanol.
Yield: 194 mg (60%). m.p.: 257-259°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ = 1.03-1.06 (m, 3H), 1.59 (s, 1H), 1.78-1.87 (m, 2H), 2.41 (d, J=4.4 Hz, 2H), 4.08-4.12 (m, 2H), 7.12-7.15 (m, 1H), 7.22-7.27 (m, 2H), 7.27-7.31 (m, 1H), 7.48-7.52 (m, 2H), 7.52- 7.57 (m, 2H), 7.77-7.79 (dd, J=8.4, 1.1 Hz, 2H). 13C-NMR (100 MHz, CDCl3): δ = 24.7, 120, 124.4, 129.1, 139, 169.6 (CO). IR (KBr): ν̃ (cm- 1) = 2962, 2930, 2875, 2362, 2341, 1643 (CO), 1595, 1549, 1482, 1442, 1379, 1338, 1274, 1158, 988, 775, 752, 696, 678, 620. UV λmax (MeOH): 198, 273 nm. GC-MS (70 eV) m/z (%): 321 (15) [M+], 277 (15), 249 (14), 145 (19), 118 (20), 90 (100), 77 (35), 41 (27).
(E)-N-Hydroxy-N’-phenylacetimidamide (11) (Method A): Hydroxylamine hydrochloride (1.05 g, 15.0 mmol) and sodium carbonate (1.5 g, 14.2 mmol) was added to a solution of 8 (1.5 g, 10 mmol) in dioxane (40 mL) and stirred for 48 hours at 50°C. The mixture was taken up in DCM (30 mL) and washed with water twice (2 × 20 mL). The organic layer was dried over Na2SO4. The solvent was removed in vacuo. The crude residue was subjected to flash column chromatography using a solvent mixture of ethyl acetate /hexane (4:1). The white product collected after evaporation of the solvent was then recrystallized from water to give white crystals.
(Method B): A mixture of nitroethane (0.670 mL, 12.5 mmol) and aniline (0.913 mL, 10 mmol) in PPA (20 g, 86% P2O5) was vigorously stirred. The reaction mixture was heated for 5 hours at 110°C. The mixture was cooled down to 80°C and diluted with water (50 mL) after TLC confirmed the total consumption of starting material. An aqueous solution of ammonia (20%) was used to neutralize the mixture (pH Ì´ 9), followed with heating to reflux. After filtration, the filtrate was cooled down to 0°C. The resulting crystalline precipitate was collected by suction filtration and recrystallized from water.
Yield: A) 825 mg (55%). B) 1.17 g (78%). m.p.: 120°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ = 1.97 (s, 3H), 7.06-7.07 (dd, J=8.4, 1.1 Hz, 2H), 7.12-7.15 (m, 1H), 7.29-7.34 (m, 2H). 13C-NMR (100 MHz, CDCl3): δ =16.1, 123.9, 124.7, 129.3, 139.0, 150.7 (CO). IR (KBr): ̃ (cm-1) = 3356, 3102 (OH), 3046, 2924, 1639, 1597, 1500, 1433, 1410, 1366, 1305, 1034. UV λmax (MeOH): 198, 245 nm. GC-MS (70 eV) m/z (%): 150 (15) [M+], 133 (33), 118 (30), 93 (100), 77 (74), 65 (35), 51 (24).
3-Methyl-4-phenyl-1,2,4-oxadiazol-5(4H)-one (12): Under a nitrogen atmosphere, to a solution of 11 (150 mg, 1.0 mmol) in anhydrous dioxane (10 mL) was added CDI (535.1 mg, 3.3 mmol). The reaction mixture was stirred at 50°C for 24 hours and the solvent removed in vacuo. The residue was taken up in DCM and washed three times with water. The organic layer was dried over Na2SO4 and the solvent removed using a rotary evaporator. The crude residue was purified by flash column chromatography to obtain a white solid. The solid was further purified by recrystallization using hexane/ ethyl acetate to obtain white crystals.
Yield: 106 mg (60%). m.p.: 133-135°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ=2.19 (s, 3H), 7.30-7.32 (d, J=2.9 Hz, 1H), 7.51-7.54 (m, 2H), 7.55-7.56 (m, 1H). 13C-NMR (100 MHz, CDCl3): δ=11.3, 126.8, 130.1, 130.2, 131.2, 156.2 (CO). IR (KBr): ν̃ (cm- 1) = 3062, 2926, 1766, 1595, 1502, 1445, 1420, 1310, 1168, 1080, 1004, 885, 756, 692, 623. UV λmax (MeOH): 197, 274 nm. GC-MS (70 eV) m/z (%): 176 (34) [M+], 131 (54), 91 (83), 77 (100), 64 (62), 51 (56).
3-Methyl-4-phenyl-1,2,4-oxadiazole-5(4H)-thione (13): Under a nitrogen gas, compound 11 (150 mg, 1.0 mmol) was dissolved in anhydrous dioxane (10 mL) and added to TDI (587.9 mg, 3.3 mmol). The reaction mixture was stirred at 50°C for 12 hours and the solvent removed in vacuo. The residue was taken up in DCM and washed three times with water. The organic layer was dried over Na2SO4 and the solvent removed in vacuo. The crude residue was purified by flash column chromatography to obtain a pale yellow solid, 150 mg (78% yield). The solid was further purified by recrystallization using hexane/ ethyl acetate to obtain white crystals.
Yield: 145 mg (78%). m.p.: 135-137°C. =0º (c=0.5, CDCl3)
1H-NMR (400 MHz, CDCl3): δ=2.20 (s, 3H), 7.32-7.35 (td, J=3.8, 1.8 Hz, 2H), 7.57-7.62 (m, 3H). 13C-NMR (100 MHz, CDCl3): δ = 10.4, 124.9, 127.5, 128.5, 130.4, 130.9, 132.6, 167.1 (CN), 186.7 (CS). IR (KBr): ν̃ (cm-1) = 2956, 2914, 1591, 1495, 1348, 1294, 1141. UV λmax (MeOH): 198, 274 nm. GC-MS (70 eV) m/z (%): 192 (40) [M+], 123 (56), 91 (70), 77 (100), 64 (70), 51 (56).
Kinetics studies of β-lactamases (inhibition assay)
The lyophilized enzymes were solubilized to 47.5 nM and 90 nM for TEM-1 and P99 respectively in 20 mM MOPS and 1% bovine serum albumin (BSA) at pH 7.5 and stored at -80°C as per the manufacturer’s protocol. All PBD derivatives after synthesis and characterization were solubilized to 12 mM in dimethylformamide (DMF) and stored at -20°C. The purity of all compounds (inhibitors) was validated by 1H-NMR, 13C-NMR, mass spectrometry, TLC, and HPLC. The reaction buffer prepared for use in all assays was 20 mM MOPS with 0.1% BSA adjusted to pH 7.4 and stored at 4°C. Enzyme activity quantitation was by spectrophotometric measurement of the hydrolysis of nitrocefin (NCF) at 485 nm and at 30°C (Δε=20,500 M-1 cm-1). NCF was present at 100 μM, TEM-1 was at 0.25 nM, and P99 was at 0.2 nM in 20 mM MOPS buffer, pH 7.0, with 0.1% bovine serum albumin (buffer A) in a final volume of 600 μL. Compounds 1-7 and 11-13 were used at final concentrations of 600 μM. Initial rates were monitored for 5 minutes on an Agilent Technologies Cary 8454 UV-Vis spectrophotometer which was also used for determination of the percentage inhibition and enzyme residual activity. Percentage enzyme inhibition was calculated by the formula below:
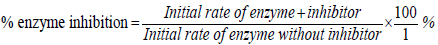
Enzyme residual activity was calculated thus:
Enzyme Residual Activity (%) =100 %−% enzyme inhibition
Molecular modeling studies
ParDOCK from SCFBio is an all-atom energy based Monte Carlo, rigid protein-ligand docking, implemented in a fully automated, parallel processing mode which predicts the binding mode of the ligand in receptor target site. The structural input data for the ParDOCK are optimized reference complex i.e., protein bound with a ligand and a candidate ligand to be docked. ParDOCK is tested on 226 proteinligand complexes [29]. Docking studies of selected PBD analogs were performed using this software. The crystal structure the TEM-1 β-lactamase was downloaded from the RCSB Protein Data Bank. All structures of the new PBD compounds were prepared using Acceryls Discovery Studio and converted to pdb format. ParDOCK was used for computational studies and determination of binding affinity energies. Discovery Studios was used to show the residues involved in hydrogen bonding, bond length and bond angles. Residues mainly involved in hydrogen bonding interaction with ligands were: Ser 45, Ser 105, Ser 230, Arg 218, Lys 209 and Ala 212. As noted in other studies, it has been shown that active site residues Ser 45, Ser 105 and Ser 230 are crucial for catalytic activity of enzyme and thus, are conserved.
X-ray crystallography
A large colorless prism was cut (0.10 × 0.15 × 0.23 mm3) and centered on the goniometer of a Rigaku Oxford Diffraction Gemini E diffractometer operating with MoKα radiation. The data collection routine, unit cell refinement, and data processing were carried out with the program CrysAlisPro [30]. The Laue symmetry and systematic absences were consistent with the monoclinic space groups I2 and I2/m. As the sample was known to be enantiomerically pure, the acentric space group, I2, was chosen to give Z=4 and Z’=1. The absolute configuration could not be determined from the anomalous dispersion effects. The structure was solved using SHELXS-2014 [31] and refined using SHELXL-2014 via Olex2 [32]. The final refinement model involved anisotropic displacement parameters for non-hydrogen atoms and a riding model for all hydrogen atoms.
Results and Discussion
All compounds designed and synthesized during the course of this research were tested for conformity to at least two of the five Lipinski’s rules for drug-likeness [32,33]. As shown in Table 1, compounds 1-6 and 10-13 adapted minimum of two rules with respect to druglikeness. Drug-likeness, as described by the Lipinski’s rule of five, is based on a number of factors and it is a qualitative concept used in drug design to ascertain how drug-like a compound is with regards to its bioavailability. Lipophilicity is the ability of a compound to be soluble in fat and usually measured experimentally using a model system or computationally as it was done in this work where it is termed cLOGP. Also, drugs which are less than 500 Daltons in size are known to be usually more efficient in terms of easy diffusion into the cell. All compounds were determined to have low to moderate cLOGP, molecular weights ranging from 150-403 Daltons, low hydrogen bond donors (HBDs), moderate hydrogen bond acceptors (HBAs), and molar refractivity ranging from 44.28-94.70.
| Lipinski rule of five parameters |
Compounds |
| 1 |
2 |
3 |
4 |
5 |
6 |
10 |
11 |
12 |
13 |
| Mass (daltons) [<500] |
216 |
232 |
257 |
403 |
231 |
259 |
325 |
150 |
176 |
192 |
| Lipophilicity (cLOGP) [<5] |
1.24 |
2.04 |
2.33 |
3.00 |
1.21 |
1.09 |
1.78 |
1.61 |
1.978 |
2.33 |
| Hydrogen bond donors [<5] |
1 |
1 |
1 |
0 |
2 |
1 |
2 |
2 |
0 |
0 |
| Hydrogen bond acceptors [<10] |
4 |
3 |
4 |
6 |
5 |
6 |
3 |
3 |
4 |
2 |
| Molar refractivity [40-130] |
59.13 |
66.72 |
75.89 |
112.96 |
62.67 |
66.34 |
94.70 |
44.28 |
48.51 |
53.33 |
Table 1: Lipinski’s rule of five data for compounds 1-6 and 10-13.
Molecular modeling studies
To gain insight the binding mode of the PBD derivatives, the select PBD derivatives (e.g., 1, 2, 4 and 6) were docked on to TEM-1 β-lactamase (PDB code: 1LI0) using ParDock software from SCFBio, India (Figure 4). Results from molecular docking revealed the interaction of ligands with different amino acid residues of the active site of TEM-1 β-lactamase as shown in Table 2 below.
| Predicted active site residues and predicted binding affinity energies |
1 |
2 |
3 |
4 |
5 |
6 |
10 |
11 |
12 |
13 |
| Binding Affinity Energies(kcal/mol) |
-5.10 |
-5.55 |
-5.81 |
-7.14 |
-6.86 |
-4.42 |
-6.51 |
-3.64 |
-3.03 |
-4.05 |
| SER45 |
2.66 |
2.71 |
3.73 |
3.36 |
- |
1.76 |
- |
2.10 |
3.48 |
3.41 |
| SER105 |
2.75 |
2.69 |
3.21 |
4.18 |
- |
1.71 |
- |
- |
3.59 |
- |
| SER210 |
- |
- |
- |
- |
1.55 |
- |
1.89 |
- |
- |
- |
| LYS48 |
- |
- |
- |
- |
2.05 |
- |
- |
- |
- |
- |
| ASN107 |
- |
- |
- |
- |
- |
- |
- |
2.48 |
2.25 |
- |
| ALA212 |
- |
- |
- |
- |
1.84 |
- |
- |
- |
- |
- |
| ARG218 |
- |
- |
- |
- |
- |
- |
1.94 |
- |
- |
- |
| LYS209 |
- |
- |
2.87 |
- |
- |
- |
1.95 |
- |
- |
- |
| GLU141 |
- |
- |
- |
- |
- |
- |
- |
2.14 |
- |
- |
Table 2: Predicted binding affinity, hydrogen bond distances, and active site residues of TEM-1 β-lactamase interacting with ligands.
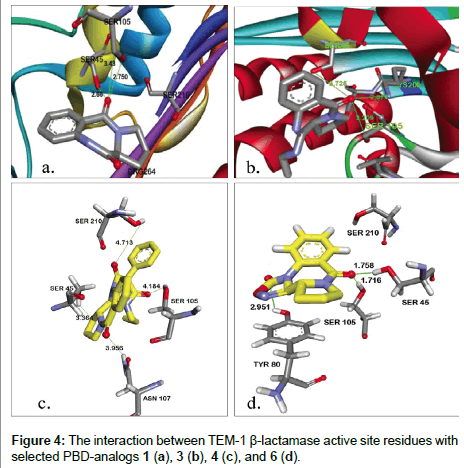
Figure 4: The interaction between TEM-1 β-lactamase active site residues with selected PBD-analogs 1 (a), 3 (b), 4 (c), and 6 (d).
From the data in Table 2, possible interactions between some of the PBD derivatives and the active site residues of the TEM- β-lactamase crystal structure were observed. Ser70 (here seen as Ser45) which is one of the most important catalytic active site residue for β-lactam hydrolysis as well as other Serine active site residues (Ser105, Ser210) were found to be interacting with the PBD derivatives docked. All PBDs had good predicted binding affinity energies ranging from -5.10 to -7.14 kcal/mol. Conserved active site residues were observed to be interacting/forming hydrogen bonds at distances mostly <3 Å as shown in Figure 4a-4d. Unlike our initial expectation, a careful supervision of effective hydrogen bondings within the active site of TEM-1 disclosed that the projected electrophilic site of PBD-ligands (sec-amide) is faced away from the cavity. Therefore, prymidinone 10 and oxadiazolinones 12 and 13 were designed to evade possible rejection of ligands from the enzyme recognition sites because of steric hindrance (Figure 5).
Figure 5: The interaction between TEM-1 β-lactamase active site residues with 12.
New unprotected ligands were created by removing of the pyrrolidine from the PBD parent molecule. A similar approach to PBD-analogs was applied to navigate the enzyme-ligand tightness of prymidinone and oxadiazolinones; thereafter, we reevaluated the binding affinity and hydrogen bonding within the enzyme active site (Table 2). Although compound 10 exhibits the highest binding energy, however, its effective hydrogen bonding with key γ-OH of Ser-45 is absent, most likely because of remarkable repulsive forces resulted from two phenyls and one n-propyl groups. In contrast, ligand 12 shows satisfactory hydrogen bonding with Ser-45 but displays lack of adequate binding affinity. Based on the computer-based molecular modeling results, all proposed PBD (1-7) and N-phenylacetamide (8-13) derivatives were synthesized either with a new method or a modification of previously reported procedures.
Chemistry
PBD natural product (1) (from Isatis indigotica [34]) was produce as the starting material for all other derivatives by refluxing isatoic anhydride with (L)-proline in DMF, following literature procedures (Scheme 1) [35,36]. Dilactam 1 was converted to thiolactam 2 in high yield (88%) [37]. The monothiolactam 2 was then reacted with an amine (n-propylamine) in the presence of mercury(II)chloride (HgCl2) to the cyclic amidine 3 in 78% yield.
Scheme 1: Reagents and conditions: (a) L-proline, DMF, 155°C, 5 h, 93.6%; (b) Lawesson’s reagent, THF, rt, 24 h, 88.0%; (c) R-NH2, HgCl2, THF, reflux, 1 h, 78% (d) magic ester, 190-220°C, 10 min, 75%; (e) NH2OH.HCl, K2CO3, EtOH (abs.), rt, 24 h, 94%; (f) CDI, dioxane (anhy.), reflux, 12 h, 88%; (g) TDI, dioxane (anhy.), reflux, 12 h, 90%. CDI: carbonyldiimidazole, tDI: thionyldiimidazole, Tcp: trichlorophenyl, Ph: phenyl.
Pyrimidine-annulated pyrrolobenzodiazepine 4 was formed by reacting of the amidine 3 with bis(2,4,6-trichlorophenyl)-2- phenylmalonates using a neat reaction (performed in a Zincke apparatus) with the leaving group of 2,4,6-trichlorophenol formed during the reaction being progressively distilled off. In general, the reaction of N, N´-disubstituted amidines with bis(2,4,6-trichlorophenyl) malonates has been shown to result in the formation of pyridinium-4-olates. Ring closure by the loss of two molecules of trichlorophenol through a ketene intermediate is proposed to be the possible explanations for the syntheses of these compounds mechanistically [38].
A modification of the protocol used by Rekowski et al. and Bartsch et al. was employed for the synthesis of oxime 5 [39,40]. Compound 5 was formed by a nucleophilic substitution of 2 using NH2OHâˆÂââ€Å¾Â¢HCl under basic conditions. The % yield of the reaction improved significantly from 76% reported in the literature to 94% with the change in the base from triethylamine to K2CO3. Compounds 6 and 7 were generated through carbonylation and thionylation reactions, respectively. Oxime 5 was treated with 1,1-carbonyl diimidazole (CDI) and 1,1-thionyl diimidazole (TDI) and refluxed in anhydrous dioxane for 12 hours, respectively. The change in the solvent from THF to dioxane made the reaction faster and more efficient. The reaction with THF according to literature occurs in 24 hours and produces a yield of 84% [41]. The modification leads to the formation of 6 and 7 in 12 hours with yields of 88% and 90%, respectively. Single crystals of compounds 6 and 7 which have not previously been published in literature were also produced.
To gain additional insights into the chemical structure of 6, we tried to obtain single crystals for an X-ray analysis. We were finally successful by slow evaporation of a concentrated solution of 6 in a 1:1 mixture of diisopropyl ether and ethyl acetate. The elemental cell contains one molecule of the PBD oxadiazole. The molecular structure and crystallographic numbering of 6 are shown in Figure 6.
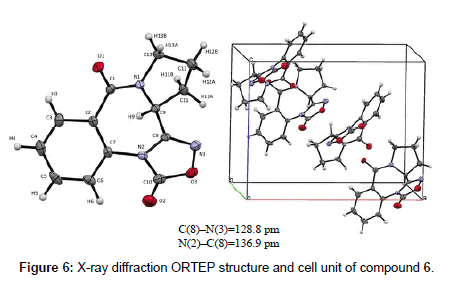
Figure 6: X-ray diffraction ORTEP structure and cell unit of compound 6.
The conformation adopted by 6 according to the X-ray diffraction ORTEP structure was a twisted conformation which is synonymous with 6:7:5 pyrrolobenzodiazepine ring systems. The seven membered ring in this structure adopts a boat arrangement which is confirmed by bond angles of N(2)-C(8)-C(9) and C(8)-N(2)-C(7) which were determined to be 119.90 (19º) and 126.35 (2º), respectively. The C(8)-N(3) bond length is 128.8 pm which corresponds to an imino C(sp2)=N(sp2) double bond. On the other hand, the N(2)-C(8) represents a single bond which has a bond length of 136.9 pm. The distinct C(8)-C(9) single bond present in the structure with a bond length of 149.5 pm excludes the formation of an optically inactive tautomer of 6. Using similar methodology to PBD-analogs, stripped compounds 8-13 were synthesized to reduce the bulkiness of the ligands (Scheme 2). Thioacetamide 8 was made by the thionation of N-phenylacetamide using Lawesson’s reagent at room temperature for 4 h [42]. Next, compound 8 was reacted with n-propyl amine in the presence of mercury(II)chloride to yield (E)-N-phenyl-N’-propylacetimidamide (9) which was a liquid with a boiling point of about 150°C. Amidine 9 was converted to primidinone 10 using the same neat reaction used for the synthesis of PBD oxo-pyrimidines 4.
Scheme 2: Reagents and conditions: (a) Lawesson’s reagent, DCM, rt, 4 h, 93.0%; (b) n-Pr-NH2, HgCl2, THF, 0°C, 2 h, 70%; (c) PPA, 105°C, 5 h, 75%; (d) magic ester, 100°C, 10 min, 60%; (e) NH2OH.HCl, Na2CO3, THF (anhy.), reflux, 24 h, 55%; (f) CDI, dioxane (anhy.), 50°C, 24 h, 60%; g) TDI, dioxane (anhy.), 50°C, 12 h, 78%.
Two different synthetic routes were employed for the synthesis of oxime 11. The first method was similar to the one used for the formation of compound 5, where 11 is formed by nucleophilic substitution of 8 with NH2OHâˆÂââ€Å¾Â¢HCL in the presence of Na2CO3 as a mild base. The yield was fairly lower (55% yield) when compared to the other synthetic procedure that have been reported in the literature for the synthesis of 11 [43]. In the second synthetic route, aniline was reacted with nitroethane in the presence of polyphosphoric acid (PPA). The reaction was quenched and neutralized after 5 hours using H2O and then NH3 (aq), respectively. Final product 11 recrystallized from the water. 1,2,4-oxadiazole-5-(thi)ones 12 and 13 were also formed through carbonylation and thionylation reactions, respectively, similar to their PBD counterparts. In these reactions, however, dioxane was used as the solvent and the reaction temperature was reduced to 50°C. Crystals of compounds 12 and 13 were also recovered from a mixture of diisopropyl ether and ethyl acetate.
Kinetics studies
To determine the residual activity (%) and percentage inhibition of the ligands, a kinetic analysis of the enzyme inhibition was carried out using TEM-1 and P99 in 20 mM of MOPS buffer. Single-dose inhibition of the enzymes was determined after 5 min incubation with inhibitory compounds in the presence of chromogenic substrate NCF. Initial deactivation rates of β-lactamases were conducted to collect the inhibition data in accordance with Michaelis-Menten enzyme kinetics for an enzyme-substrate inhibitor system (Tables 3 and 4). From the enzyme kinetics results, it was observed that all derivatives of PBD (1- 7) and N-phenylacetamide (11-13) show little to no inhibitory activity against the enzymes used in this study.
| Compounds |
Vo± SD (ΔA, s-1) × 10-4 |
Vi± SD (ΔA, s-1) × 10-4 |
Residual Activity (%) |
% Inhibition |
| 1 |
2.3721 ± 0.0179 |
1.5667 ± 0.01686 |
66.05 |
33.95 |
| 2 |
2.3721 ± 0.0179 |
2.5101 ± 0.08144 |
100 |
NA |
| 3 |
2.3721 ± 0.0179 |
2.6475 ± 0.04724 |
100 |
NA |
| 4 |
2.3721 ± 0.0179 |
2.0864 ± 0.01605 |
87.96 |
12.04 |
| 5 |
2.3721 ± 0.0179 |
1.5617 ± 0.01740 |
65.83 |
34.17 |
| 6 |
2.3721 ± 0.0179 |
2.1298 ± 0.02806 |
89.79 |
10.21 |
| 7 |
2.3721 ± 0.0179 |
2.1795 ± 0.02172 |
91.88 |
9.22 |
| 11 |
3.7575 ±0.02961 |
4.2356 ± 0.052340 |
100 |
NA |
| 12 |
3.7575 ±0.02961 |
4.6721 ± 0.035013 |
100 |
NA |
| 13 |
3.7575 ±0.02961 |
3.3132 ± 0.064359 |
88.18 |
11.82 |
Final concentration of TEM-1 and P99=0.25 nM; Final concentration of P99=0.20 nM. Substrate (NCF)=12 μL; Triton X-100=0.01%; Buffer=0.1% BSA in MOPS (0.02 M, pH 7.5). Inhibitor final concentration in 3% DMF=600 μM. NA=No activity
Table 3: Residual Activity (%) and percent inhibition data for TEM-1 after incubation PBD (1-7) and phenyl acetamide (11-13) derivatives for 5 min at 30°C in DMF (3%).
| Compounds |
Vo± SD (ΔA, s-1) × 10-4 |
Vi± SD (ΔA, s-1) × 10-4 |
Residual Activity (%) |
% Inhibition |
| 1 |
2.5743 ± 0.01647 |
3.9885 ± 0.04783 |
100 |
NA |
| 2 |
2.5743 ± 0.01647 |
3.802 ± 0.08103 |
100 |
NA |
| 3 |
2.5743 ± 0.01647 |
3.4348 ± 0.01447 |
100 |
NA |
| 4 |
2.5743 ± 0.01647 |
2.2329 ± 0.01290 |
86.74 |
13.36 |
| 5 |
2.5743 ± 0.01647 |
2.6279 ± 0.03439 |
100 |
NA |
| 6 |
2.5743 ± 0.01647 |
2.2965 ± 0.02968 |
89.24 |
10.76 |
| 7 |
2.5743 ± 0.01647 |
2.4733 ± 0.02748 |
96.08 |
3.92 |
| 11 |
4.4748 ± 0.02435 |
4.5517 ± 0.035475 |
100 |
NA |
| 12 |
4.4748 ± 0.02435 |
4.6837 ± 0.041379 |
100 |
NA |
| 13 |
4.4748 ± 0.02435 |
5.0231 ± 0.028540 |
100 |
NA |
Final concentration of TEM-1 and P99=0.25 nM; Final concentration of P99=0.20 nM; Substrate (NCF)=12 μL; Triton X-100=0.01%; Buffer=0.1% BSA in MOPS (0.02 M, pH 7.5). Inhibitor final concentration in 3% DMF=600 μM; NA=No activity
Table 4: Residual Activity (%) and percent inhibition data for P99 after incubation of PBD (1-7) and phenyl acetamide (11-13) derivatives for 5 min at 30°C in DMF (3%).
Compounds 1, 4, 5 and 6 exhibit weak inhibitory activity against TEM-1 while compounds 4, 6, and 7 display very low inhibitory activities against P99. Compound 5 had the highest percentage inhibition of 34.17% for TEM-1 β-lactamase but had no effect on P99 at a final concentration of 600 μM. Compounds 4, 6, and 7 also showed insignificant percentage inhibition versus TEM-1 in the amount of 12.04, 10.21, and 9.22%, respectively. This relative low inhibition was replicated in P99 enzyme with values of 13.36, 10.76, and 9.21% for compounds 4, 6, and 7, respectively. The low potency associated with many of the ligands in this study may be due to limited solubility and depleted polarity of the inhibitors in the buffer solution used for the enzyme kinetics reactions, and hence leading to poor delivery of the ligands to the active site.
There is also a possibility that steric interference being another setback preventing the active part of the molecules from the optimal interaction with the active site residues of the enzymes. We assumed that incorporation of polar functional groups such as CO2H, SO3H, and OH, along with removal of the bulky group repulsions would be essential to improve the activity of the inhibitors versus the enzymes.
Molecular docking results showed the possibility of pyrimidinone 11 and oxadiazolinones 12 and 13 as potential non-β-lactam β-lactamase inhibitors due to their sufficient interaction with the enzymes’ active site residues and predicted binding affinity. Ligand 12 displayed better interaction in terms of the number of active site residues, as well as hydrogen bonding forces (Figure 5). As a result, derivatives 11-13 were synthesized to relief the possible steric hindrance and likely increase the electrophilicity and optimal interaction between active site residues and the inhibitors. Unfortunately, after in vitro assays, only 13 showed a weak activity against TEM-1 with a percentage inhibition of 11.82% at a final concentration of 600 μM.
Based on the poor inhibitory activities of the PBD and N-phenylacetamide derivatives observed during the enzyme kinetics experiment, we proposed some possible explanation for these poor results. The poor inhibitory activities of the ligands could have been as a result of the formation of second resonance structure (A↔B, aromatization) which may significantly reduce the activity of carbonyl group and prevent the construction of tetrahedral intermediate in deacylation step (Scheme 3). We believe that elimination of aromaticity by hydrogenation may result in revival of the activity. Thus, structures 14-17 are suggested following our new strategy.

Scheme 3: Proposed explanation for low activity of the designed ligands and suggested new compounds.
Also, substituting the bulky groups with smaller and more polar functional groups will possibly boost the optimal interactions and consequently will increase the efficacy of designed molecules. The search for more elaborated chemotypes through fragment-based design which may have better activity as well as stronger affinity, followed by synthesis and further in vitro evaluation against class A and C serine β-lactamases is currently under investigation.
Acknowledgements
The authors acknowledge the Department of Chemistry and The School of Graduate Studies at ETSU. We are also grateful for the financial support of the ETSU Office of Research and Sponsored Programs Administration (ORSPA).
11214
References
- Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, et al. (2008) The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis46:155-164.
- World Health Organization (2012) The evolving threat of antimicrobial resistance: Options for action, 2012.
- Pratt RF (2002) Functional evolution of the serine β-lactamase active site. J Chem Soc PerkinTrans2:851-861.
- Zervosen A, Sauvage E, Frère JM, Charlier P, Luxen A (2012) Development of new drugs for an old target-the penicillin binding proteins. Molecules17:12478-12505.
- Bush K (1988) β-Lactamase inhibitors from laboratory to clinic. Clin Microbiol Rev 1:109-123.
- Gill EE, Franco OL, Hancock REW (2015) Antibiotic adjuvants: Diverse strategies for controlling drug-resistant pathogens. Chem Biol Drug Des85:56-78.
- Drawz SM, Bonomo RA (2010) Three decades of β-lactamase inhibitors. Clin Microbiol Rev23:160-201.
- Pucci MJ, Bush K (2013) Investigational antimicrobial agents of 2013. Clin Microbiol Rev 26: 792-821.
- Stachyra T, Pechereau MC, Bruneau JM, Claudon M, Frere JM, et al. (2010) Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-beta-lactam beta-lactamase inhibitor. Antimicrob Agents Chemother 54:5132-5138.
- Garber K (2015) A β-lactamase inhibitor revival provides new hope for old antibiotics. Nat Rev Drug Discov14:445-447.
- Ge Y, Difuntorum S, Touami S, Critchley I, Bürli R, et al. (2002) In vitro antimicrobial activity of GSQ1530, a new heteroaromatic polycyclic compound. Antimicrob Agents Chemother46:3168-3174.
- Hadjivassileva T, Stapleton PD, Thurston DE (2007) Interactions of pyrrolobenzodiazepine dimers and duplex DNA from methicillin-resistant Staphylococcus aureus. Int J Antimicrob Agents 290:672-678.
- Doyle M, Feuerbaum EA, Fox KR(2009) Response of Staphylococcus aureus to subinhibitory concentrations of a sequence-selective, DNA minor groove cross-linking pyrrolobenzodiazepine dimer. J Antimicrob Chemother64:949-959.
- Denny WA (2001) DNA minor groove alkylating agents. Curr Med Chem8:533-544.
- Hadjivassileva T, Thurston DE, Taylor PW (2005) Pyrrolobenzodiazepine dimers: novel sequence-selective, DNA-interactive, cross-linking agents with activity against Gram-positive bacteria. J Antimicrob Chemother 56:513-518.
- Jackson PJ, James CH, Jenkins TC, Rahman KM, Thurston DE (2014) Computational studies support the role of the C7-sibirosamine sugar of the pyrrolobenzodiazepine (PBD) sibiromycin in transcription factor inhibition. ACS Chem Biol9:2432-2440.
- Puvvada MS, Forrow SA, Hartley JA, Stephenson P, Gibson I, et al. (1997) Inhibition of bacteriophage T7 RNA polymerase in vitro transcription by DNA-binding pyrrolo[2,1-c][1,4]-benzodiazepines. Biochemistry36: 2478-84.
- Leimgruber W, Stefanovic V, Schenker F, Karr A, Berger J (1965) Isolation and characterization of anthramycin, a new antitumor antibiotic. J Am Chem Soc87:5791-5793.
- Arima K, Kosaka M, Tamura G (1972)Studies on tomaymycin, a new antibiotic. I. Isolation and properties of tomaymycin. J Antibiot (Tokyo)25:437-444.
- Brazhnikova MG, Konstantinova NV, Mesentsev AS (1972)Sibiromycin: isolation and characterization. J Antibiot (Tokyo)25:668-673.
- Cargill C, Bachmann E, Zbinden G (1974) Effects of daunomycin and anthramycin on electrocardiogram and mitochondrial metabolism of the rat heart. J Natl Cancer Inst 53:481-486.
- Lubawy WC, Dallam RA, Hurley LH (1980)Protection against anthramycin-induced toxicity in mice by coenzyme Q10. J Natl Cancer Inst64:105-109.
- Schmidt A, Shilabin AG, Namyslo JC, Nieger M, Hemmen S (2005) Pyrimidine-annulated pyrrolobenzodiazepines. A new ring system related to Aspergillus alkaloids. Eur J Org Chem1781-1789.
- Schmidt A, Lindner AS, Shilabin AG, Nieger M (2008) New derivatives and ring systems of annulated pyrrolobenzo-[1, 4]-diazepines. Tetrahedron64:2048-2056.
- Schmidt A, Shilabin AG, Nieger M (2005) Syntheses and tautomerization of amino-substituted and pyrimidine-annulated pyrrolobenzodiazepines. Heterocycles65:625-632.
- Schmidt A, Shilabin AG, Nieger M (2004) Thiazolidinone-annulated pyrrolobenzodiazepines. Syntheses and properties of a new ring system Heterocycles 63:2851-2858.
- Rahman KM, Rosado H, Moreira JB, Feuerbaum E-A, Fox KR, et al. (2012) Antistaphylococcal activity of DNA-interactive pyrrolobenzodiazepine (PBD) dimers and PBD-biaryl conjugates. J Antimicrob Chemother67:1683-1696.
- Drawz SM, Bonomo RA (2010) Three decades of β-lactamase inhibitors. Clin Microbiol Rev23:160-201.
- Gupta A, Gandhimathi A, Sharma P, Jayaram B (2007) ParDOCK: An all-atom energy based Monte Carlo docking protocol for protein-ligand complexes. Protein Pept Lett14: 632-646.
- CrysAlisPro Software System (2015) v1.171.37.35, Rigaku Oxford Diffraction, 2015, Rigaku Corporation, Oxford, UK.
- Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H (2009) OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst42:339-341.
- Lipinski CA, Lombardo, F, Dominy BW, Feeney PJ(2001) Experimental-computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3-26.
- Lipinski CA(2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol1: 337-341.
- Brazhnikova MG, Konstantinova NV, Mesentsev AS (1972) Sibiromycin: isolation and characterization. J Antibiot (Tokyo) 25:668-673.
- Kamal A (1991)Enzymic approach to the synthesis of the pyrrolo[1,4] benzodiazepine antibiotics. J Org Chem 56:2237-2240.
- Wright Jr WB, Brabander HJ, Greenblatt EN, Day IP, Hardy Jr RA (1978) Derivatives of 1,2,3,11a-tetrahydro-5H-pyrrolo[2,1-c][1,4]benzodiazepine-5,11(10H)-dioneas anxiolytic agents.J Med Chem 21:1087-1089.
- Kamal A, Howard PW, Reddy BSN, Reddy BSP (1997) Thurston DE. Synthesis of pyrrolo[2,1-c][1,4]benzodiazepine antibiotics: Oxidation of cyclic secondary amine with TPAP. Tetrahedron53:3223-3230.
- Shilabin AG (2005) Seven-membered ring mesomeric betaines: From anti-Hückel aromatics to model compounds of the pyrrolobenzodiazepine alkaloids-Circumdatin A and B. Ph.D. dissertation. Technischen Universität Clausthal, Clausthal-Zellerfeld, 2005.
- Rekowski MW, Pyriochou A, Papapetropoulos N, Stöbel A, Papapetropoulos A, et al. (2010) Synthesis and biological evaluation of oxadiazole derivatives as inhibitors of soluble guanylyl cyclase. Bioorg Med Chem 18:1288-1296.
- Bartsch H, Erker T, Neubauer G (1989)Untersuchungen zur Synthese neuertricyclischer Heterocyclen aus 1,4-Benzoxazin- und 1,4-Benzothiazin-3-oximen (Studien zur Chemie O,N- und S,N-haltigerHeterocyclen, 7. Mitt.). Monatsh Chem 120:81-84.
- Koduri ND, Wang Z, Cannell G, Cooley K, Lemma TM, et al. (2014) Enaminones via ruthenium-catalyzed coupling of thioamides and α-diazocarbonyl compounds. J Org Chem79:7405-7414.
- Aksenov AV, Smirnov AN, Aksenov NA, Bijieva AS, Aksenova IV, et al. (2015) Benzimidazoles and benzoxazoles via the nucleophilic addition of aniline to nitroalkanes. Org Biomol Chem 13:4289-4295.


