Tong Dai*
Division of Hematology and Oncology, Weill Cornell Medical College, 1305 York Avenue, USA
*Corresponding Author:
Tong Dai
Division of Hematology and Oncology, WeillCornell Medical College, 1305 York Avenue, 12th Floor, New York, NY 10021
USATel: 646-962-6200
Fax: 646-962-1607
E-mail: tod9013@med.cornell.edu
Received Date: May 08, 2015; Accepted Date: June 15, 2015; Published Date: June 19, 2015
Citation: Dai T. Targeting MET in Cancer: Obstacles and Potentials. Transl Biomed. 2015, 6:1. DOI: 10.21767/2172-0479.100007
HGF-MET Pathway
MET (c-Met), also known as hepatocyte growth factor (HGF) receptor, is a tyrosine kinase receptor (RTK) with multiple downstream effects including regulation of cell survival and migration of epithelial and myogenic precursor cells [1]. Upon ligand-receptor binding, phosphorylation of MET Y1234/Y1235 in the kinase domain and Y1349/Y1356 in the multi-substrate docking site facilitate binding of SH2-containing proteins and activates downstream signaling pathways including mitogenactivated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and phospholipase Cγ (PLCγ) [2-5] (Figure 1). An additional phosphorylation site in the juxtamembrane domain of MET, Y1003, modulates receptor down-regulation via ubiquitination and degradation by serving as a direct binding site for the Cbl E3- ligase tyrosine kinase binding domain [6-7].
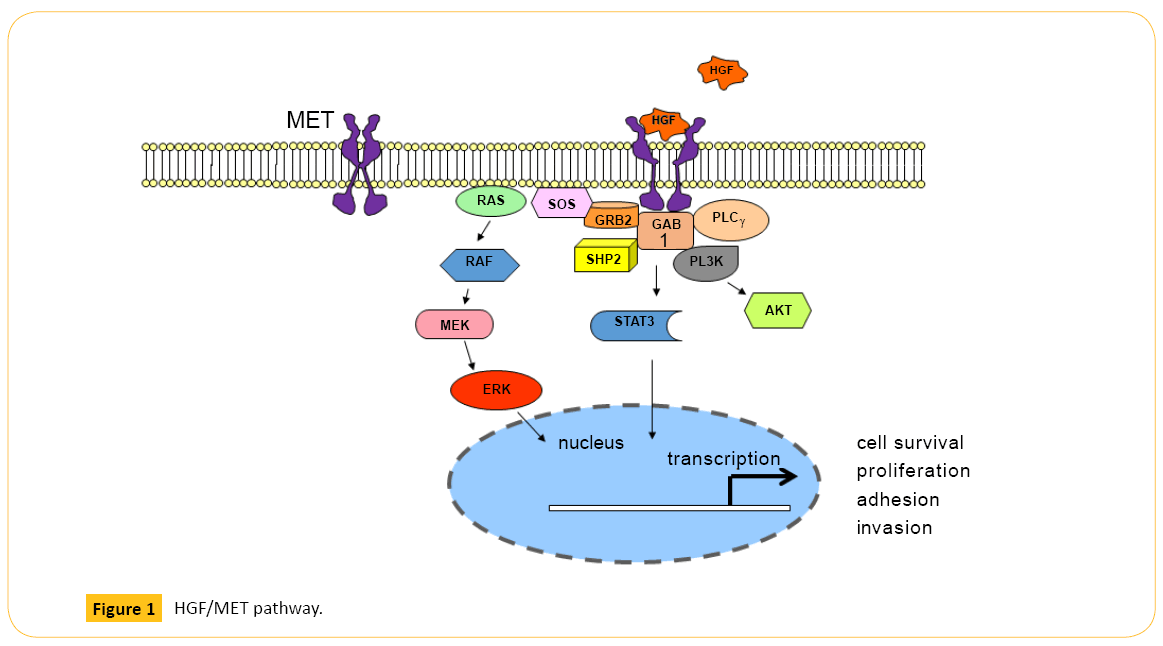
Figure 1: HGF/MET pathway.
Role of MET in Cancer
Activated MET pathway is involved in tumorigenesis and metastasis in a variety of cancer types including lung, gastric, and colon cancer [8,9]. Large scale RNA sequencing study of lung adenocarcinoma, demonstrated somatic mutations in MET as cancer driver [10]. In a cohort of 262 lung cancer patients that included predominantly non-small cell lung cancer (NSCLC) and only 2 SCLC, all instances of MET activation occurred in adenocarcinomas [11]. The prevalence of MET gene amplification and splice mutations were 1.4% and 3.3%, respectively. All 7 mutations were deletions of nucleotides 3075 to 3215 within exon 14, resulting in a 47 amino-acid deletion (L964 through D1010) in the juxtamembrane domain. Deletion of the juxtamembrane domain led to the loss of Cbl E3-ligase binding, decreased ubiquitination/degradation, and accumulation of MET protein. As a consequence, phospho-Met and downstream MAPK activation is sustained upon ligand stimulation, driving oncogenic process [12]. Exon 14 skipping was also reported as cancer driver in other studies of lung adenocarcinoma, and TCGA data from 230 resected lung adenocarcinomas revealed exon 14 skipping in 4% of cases [10,13-14]. Similarly, aberrant MET activation in gastric cancer occurs through gene amplification/overexpression and upregulation of stromal HGF ligand production [15]. In colorectal cancer (CRC), MET gene amplification is associated with advanced disease and liver metastasis which is the leading cause of CRC-related mortality [16,17].
MET activation as resistance mechanism
MET is also associated with resistance to therapies targeting other growth factor pathways [18]. For example, MET amplification was detected in 4 of 18 (22%) lung cancer that became resistant to EGFR tyrosine kinase inhibitors (TKIs) gefitinib or erlotinib [18]. Specifically, amplification of MET causes gefitinib resistance by driving ERBB3 (HER3)-dependent activation of PI3K. In another study of lung adenocarcinoma, MET was amplified in tumors from 9 of 43 (21%) patients with acquired resistance but in only 2 tumors from 62 untreated patients (3%) [19]. In human lung adenocarcinomas harboring EGFR mutations, T790M is responsible for approximately half of cases of acquired resistance to EGFR TKIs [20]. Interestingly, this study also showed MET amplification coexists with EGFR T790M mutation in 4 out 10 resistant tumors [19]. More recent study with 155 patients who became resistant to EGFR TKIs showed 63% acquired T790M mutations, MET and HER2 amplification was seen in 5% and 13%, respectively [21]. The study did not detect any acquired mutations in common cancer gene PIK3CA, AKT1, BRAF, ERBB2, KRAS, MEK1, or NRAS. In BRAF mutated melanoma, stromal cell secretion of HGF resulted in MET activation of the HGF receptor MET, conferring resistance to RAF inhibition [22].
Preclinical data shows that similar MET-mediated resistance mechanisms occur with anti-HER2 in breast cancer and gastric cancer cell lines [23]. In a study of 7 patients with KRAS wildtype metastatic CRC who initially responded to EGFR monoclonal antibody (mAb) panitumumab- or cetuximab-based treatment and then relapsed, 3 patients developed MET amplification [24]. More recent studies have identified a critical role of MET in anti-angiogenesis therapy-associated acceleration of tumor metastasis, and MET blockade significantly attenuated tumor metastasis to local lymph nodes and distant organs in breast cancer, melanoma, and pancreatic neuroendocrine tumor models [25,26].
Clinical trials targeting MET
In a phase 2 study of tivantinib, a selective, non-ATP competitive, small-molecule inhibitor of MET, as 2nd and 3rd line treatment of metastatic gastric cancer, no objective response was observed, and median progression-free survival (PFS) was only 43 days [27]. However, the data from this trial should be interpreted with caution as later studies suggested tivantinib is not a MET inhibitor, but instead a tubulin inhibitor [28].
Onartuzumab (MetMAb) is a monovalent, humanized MET mAb, specifically designed to avoid agonistic activity that may occur when a bivalent antibody binds two MET molecules (Figure 2). Onartuzumab blocks HGF-induced MET dimerization and activation of the intracellular kinase domain. Phase 2 study in recurrent NSCLC showed MET-positive patients (n=66) treated with erlotinib plus onartuzumab showed improvement in both PFS (harzad ratio (HR) 0.53; P=0.04) and overall survival (OS) (HR 0.37; P=0.002) [29]. Despite this encouraging results, addition of onartuzumab to erlotinib did not improve median OS (6.8 vs 9.1 months), median PFS (2.7 vs 2.6 months), or overall response rate (8.4% vs 9.6%) in previously treated stage IIIb or IV NSCLC in the phase 3 trial [30]. The most frequent adverse events that were higher in the combination arm were peripheral edema, hypoalbuminemia, back pain, dyspnea, nausea, acneiform dermatitis, and rash.
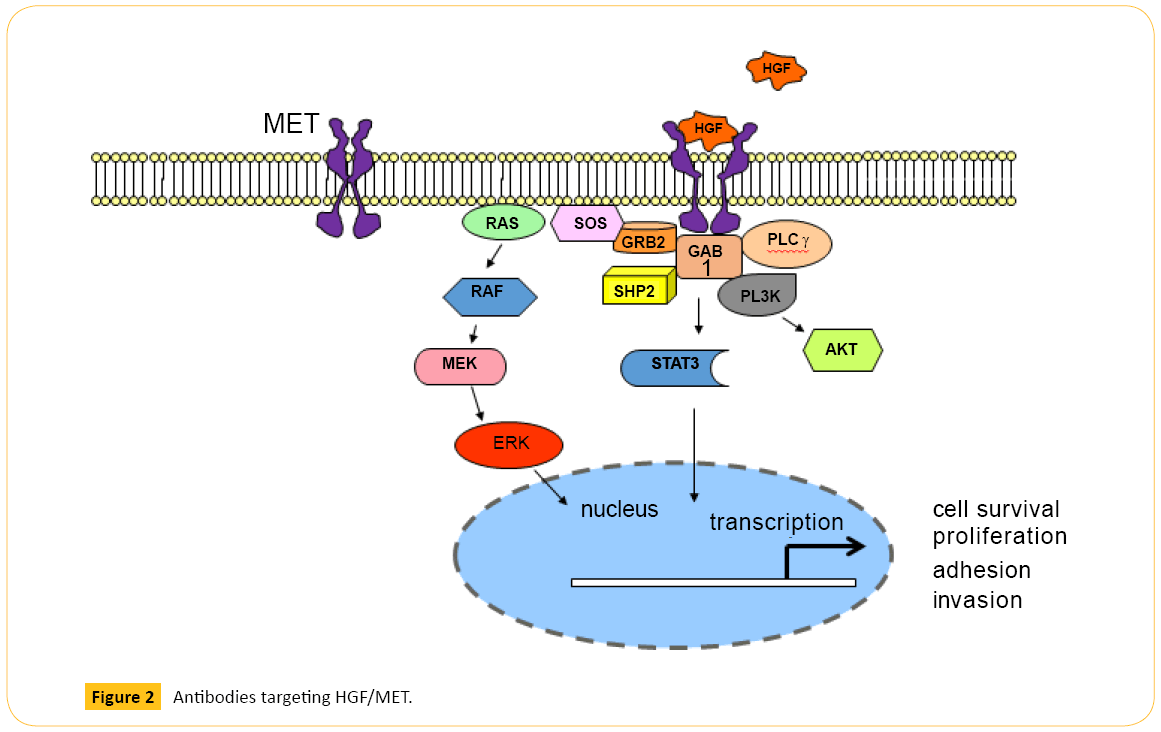
Figure 2: Antibodies targeting HGF/MET.
The results of onartuzumab in gastric cancer was also disappointing. Although a complete response of two years has been reported in a patient with metastatic gastric cancer when treated with onartuzumab on a phase 1 study [31], phase 2 study reported on 2015 GI ASCO showed addition of onartuzumab to mFOLFOX6 (fluorouracil, leucovorin, and oxaliplatin) in metastatic gastroesophageal adenocarcinoma did not improve PFS in either an unselected population or in MET-positive patients as defined by IHC [32].
Rilotumumab is a new fully humanized mAb against HGF that prevents its binding to the MET receptor (Figure 2). A recently reported double-blind phase 2 study randomly assigned 121 previously untreated patients (1:1:1) with advanced gastric or gastroesophageal junction (GEJ) cancer to receive rilotumumab 15 mg/kg, rilotumumab 7.5 mg/kg, or placebo, plus ECX (epirubicin, cisplatin, capecitabine) [33]. Median PFS was 5.1 months (P=0.164 vs. placebo group) in the rilotumumab 15 mg/kg group, 6.8 months (P=0.009) in the rilotumumab 7.5 mg/kg group, 5.7 months (P=0.016) in both rilotumumab groups combined, and 4.2 months in the placebo group. Median overall survival was longer in the combined rilotumumab groups than in the placebo group, although it is not statistically significant. In addition, in patients with MET-positive tumor (IHC or FISH confirmed), median OS was prolonged when treated with combined rilotumumab and ECC vs. ECX alone (10.6 vs. 5.7 months). This is the first randomized study of an agent targeting the HGF–MET pathway in gastric and GEJ adenocarcinoma that prolonged PFS when combined with chemotherapy in MET-high tumors. However, the phase 3 study RILOMET-1 comparing ECX plus rilotumumab (15 mg/kg) vs. placebo was recently halted due to increased deaths in the rilotumumab arm (NCT01697072).
Small molecular MET inhibitors have not shown survival benefit in late phase studies, either. In a phase 1 study, oral MET kinase inhibitor AMG 337 showed response in a subset of patients with MET-amplified gastroesophageal tumors [34], but phase 2 study in patients with MET-amplified tumors was stopped after interim analysis suggested futility.
Mechanism of resistance to MET targeted therapy
The reason multiple late phase clinical trials targeting HGF/ MET fail seems to be drug design and/or patient selection. Onartuzumab was designed to block HGF-MET interaction by targeting the beta subunit of extracellular Sema domain of MET which is required for HGF binding but may not be necessary for MET dimerization [35]. MET-amplified tumor cells normally exhibit ligand-independent, constitutive MET activation [36]. Thus, in cancers driven by MET amplification/overexpression or activating mutations, MET activation and downstream signaling is unlikely to be fully blocked by drugs solely targeting HGF-MET binding. Furthermore, it was recently shown that microenvironmentderived HGF can overcome sensitivity to anti-MET drugs [36]. Similarly, HGF mAb such as rilotumumab alone may not be able to sufficiently attenuate MET signaling in HGF-independent tumors to deliver clinical benefit. In addition, most recent late phase trials recruited MET-high patients. MET-high lung or gastric cancers determined by IHC 2+ or 3+ likely carry MET amplification and are probably HGF-independent, which may explain why they did not respond to therapy that only target HGF-MET binding.
Crosstalk of MET with EGFR and ERBB2 has emerged as an important mechanism of cancer progression and resistance to therapy [9,37]. MET pathway also interacts with WNT signaling. WNT–β-catenin stimulate MET transcription in colon cancer cell lines, and MET and integrin α3β1 signaling in turn regulate the transcription of WNT7B [38]. Furthermore, HGF stimulate LRP5/6 phosphorylation and promotes WNT signaling in the kidney [39]. In liver and bladder cancer cells, HGF/SF induces β-catenin–TCF nuclear translocation of target genes, suggesting cooperation between oncogenic pathways [40,41]. These crosstalk may also contribute to the lack of clinical efficacy when targeting HGF-MET pathway alone.
Strategy to successfully target MET
It was postulated that MET inhibition may be more clinically efficacious when used as additional therapy to block resistance pathways
activated after use of other targeted agents rather than being used alone or as 1st line treatment [9,42]. As discussed above, HGF-independent MET signaling may explain the lack of response in patients treated with mAbs which target HGFMET binding. A novel humanized bivalent anti-MET antibody, LY2875358, was recently developed to overcome this limitation (Figure 2). In addition to blocking HGF-MET binding and HGF-induced MET phosphorylation and cell proliferation, LY2875358 induces internalization and degradation of MET that inhibits cell proliferation and tumor growth in models where MET is constitutively activated. Moreover, LY2875358 has potent antitumor activity in both HGF-dependent and HGFindependent (MET-amplified) xenograft tumor models [43]. Given its unique design and mechanism of action, LY2875358 has a potential to be successful. Currently, a randomized phase 2 study is evaluating LY2875358 plus erlotinib vs. erlotinib alone as 1st line therapy in metastatic NSCLC who have disease control after 8-week lead-in with erlotinib (NCT01897480). However, if patients acquire EGFR T790M mutation on erlotinib, blocking MET pathway without switching from erlotinib to a new EGKFR TKI with T790M activity is unlikely to be sufficient. As discussed earlier, incidence of acquiring T790M mutation on EGFR TKIs is high, and MET amplification can coexist with T790M mutation [19,21].
Another very promising novel MET-targeting agent is a mAb, KTN0216, which inhibits both HGF-stimulated MET activation and HGF-independent MET kinase activation due to MET amplification/overexpression [44]. In addition to inhibiting HGF binding to MET, KTN0216 induces MET protein degradation (Figure 2). KTN0216 treatment of tumor bearing mice achieved potent and prolonged suppression of tumor growth in a U87MG glioblastoma xenograft model. More strikingly, KTN0216 led to tumor regression with tumors remaining below detection for over 90 days post last dosing using the SNU5 gastric cancer xenograft model.
Recently, HGF-neutralizing antibody ficlatuzumab was shown to sensitize MET-amplified tumors to MET-targeted agents in animal model, providing proof of concept that combined inhibition of MET and HGF may represent a potentially successful strategy [36].
Using a different approach, Hu et al reported two new types of tetra-specific antibodies that recognize EGFR, HER2, HER3, and VEGF, not only inhibited signaling mediated by these receptors but unexpectedly also disrupted HER-MET crosstalk [45]. When compared with two-in-one antibodies and a series of bispecific antibodies in multiple tumor models, they were far more effective in inhibiting the growth of anti-HER-resistant cancer cells, which exhibited elevated levels of MET activation both in vitro and in vivo. Paik et al recently demonstrated cabozantinib, an inhibitor of MET, VEGFR2, and RET, achieved partial response in a lung adenocarcinoma patient with MET exon 14 skipping mutation, suggesting simultaneous inhibition of MET and other pathway may overcome resistance to targeted therapy (unpublished data). This observation raised the hope that simultaneous inhibition of interacting/bypassing pathways in addition to HGF/MET may yield clinically meaningful result. In fact, a phase 1b/2 trial is exploring combination strategy and studying the combination of VEGFR2 mAb ramucirumab and LY2875358 in advanced cancer (NCT02082210).
Summary
Aberrant HGF-MET pathway is an oncogenic driver and is responsible for resistance in several cancer types. Strategies targeting this pathway have not been shown to prolong survival in advanced cancers. Rational design of novel agents targeting both HGF-dependent and HGF-independent MET activation, careful selection of patients for clinical trials, and development of biomarkers are the key for future success in targeting MET pathway. Further investigation to explore complex interaction and crosstalk between MET and other oncogenic pathways is imperative to elucidate its role in cancer and resistance, and may reveal novel, effective strategy using combined inhibition of MET and other pathways.
6605
References
- Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, et al. (1984) Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 311: 29-33.
- Ferracini R,Longati P, Naldini L, Vigna E, Comoglio PM (1991) Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase.J BiolChem 266: 19558-19564.
- Ponzetto C,Bardelli A, Zhen Z, Maina F, dallaZonca P, et al. (1994) A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family.Cell 77: 261-271.
- Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J, et al. (1996) Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis.Nature 384: 173-176.
- Birchmeier C,Birchmeier W, Gherardi E, VandeWoude GF (2003) Met, metastasis, motility and more.Nat Rev Mol Cell Biol 4: 915-925.
- Peschard P, Fournier TM, Lamorte L, Naujokas MA, Band H, et al. (2001) Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein.Mol Cell 8: 995-1004.
- Peschard P,Ishiyama N, Lin T, Lipkowitz S, Park M (2004) A conserved DpYR motif in the juxtamembrane domain of the Met receptor family forms an atypical c-Cbl/Cbl-b tyrosine kinase binding domain binding site required for suppression of oncogenic activation.J BiolChem 279: 29565-29571.
- Boccaccio C,Comoglio PM (2006) Invasive growth: a MET-driven genetic programme for cancer and stem cells.Nat Rev Cancer 6: 637-645.
- Gherardi E,Birchmeier W, Birchmeier C, VandeWoude G (2012) Targeting MET in cancer: rationale and progress.Nat Rev Cancer 12: 89-103.
- Seo JS,Ju YS, Lee WC, Shin JY, Lee JK, et al. (2012) The transcriptional landscape and mutational profile of lung adenocarcinoma.Genome Res 22: 2109-2119.
- Onozato R,Kosaka T, Kuwano H, Sekido Y, Yatabe Y, et al. (2009) Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers.J ThoracOncol 4: 5-11.
- Kong-Beltran M,Seshagiri S, Zha J, Zhu W, Bhawe K, et al. (2006) Somatic mutations lead to an oncogenic deletion of met in lung cancer.Cancer Res 66: 283-289.
- Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma.Nature 511: 543-550.
- Dhanasekaran SM,Balbin OA, Chen G, Nadal E, Kalyana-Sundaram S, et al. (2014) Transcriptome meta-analysis of lung cancer reveals recurrent aberrations in NRG1 and Hippo pathway genes.Nat Commun 5: 5893.
- Konturek PC, Konturek SJ, Sulekova Z, Meixner H, Bielanski W, et al. (2001) Expression of hepatocyte growth factor, transforming growth factor alpha, apoptosis related proteins Bax and Bcl-2, and gastrin in human gastric cancer. Aliment PharmacolTher 15: 989-999.
- Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan SA, et al. (2008) c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases.Cancer Lett 265: 258-269.
- Foster JH (1984) Treatment of metastatic disease of the liver: a skeptic's view.Semin Liver Dis 4: 170-179.
- Engelman JA,Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling.Science 316: 1039-1043.
- Bean J, Brennan C, Shih JY, Riely G, Viale A, et al. (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib.ProcNatlAcadSci U S A 104: 20932-20937.
- Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, et al. (2011) Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay.Clin Cancer Res 17: 1169-1180.
- Yu HA,Arcila ME, Rekhtman N, Sima CS, Zakowski MF, et al. (2013) Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.Clin Cancer Res 19: 2240-2247.
- Straussman R,Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, et al. (2012) Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion.Nature 487: 500-504.
- Liu L, Shi H, Liu Y, Anderson A, Peterson J, et al. (2011) Synergistic effects of foretinib with HER-targeted agents in MET and HER1- or HER2-coactivated tumor cells.Mol Cancer Ther 10: 518-530.
- Bardelli A,Corso S, Bertotti A, Hobor S, Valtorta E, et al. (2013) Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer.Cancer Discov 3: 658-673.
- Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, et al. (2012) Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors.Cancer Discov 2: 270-287.
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, et al. (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis.Cancer Cell 15: 232-239.
- Kang YK,Muro K, Ryu MH, Yasui H, Nishina T, et al. (2014) A phase II trial of a selective c-Met inhibitor tivantinib (ARQ 197) monotherapy as a second- or third-line therapy in the patients with metastatic gastric cancer.Invest New Drugs 32: 355-361.
- Aoyama A, Katayama R, Oh-Hara T, Sato S, Okuno Y, et al. (2014) Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes ABC transporter-mediated drug resistance.Mol Cancer Ther 13: 2978-2990.
- Spigel DR, Ervin TJ, Ramlau RA, Daniel DB, Goldschmidt JH Jr, et al. (2013) Randomized phase II trial of Onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer.J ClinOncol 31: 4105-4114.
- David RS, Edelman MJ, Kenneth OB, Luis PAs, David SS, et al. (2014) Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebo-controlled METLung (OAM4971g) global trial, in ASCO. Chicago.
- Catenacci DV, Henderson L, Xiao SY, Patel P, Yauch RL, et al. (2011) Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence.Cancer Discov 1: 573-579.
- Manish AS, Iain Tan BH, Niall CT, Yen CJ, Alice K, et al. (2015) Randomized phase II study of FOLFOX +/- MET inhibitor, onartuzumab (O), in advanced gastroesophageal adenocarcinoma (GEC). inGI ASCO : San Francisco.
- Iveson T,Donehower RC, Davidenko I, Tjulandin S, Deptala A, et al. (2014) Rilotumumab in combination with epirubicin, cisplatin, and capecitabine as first-line treatment for gastric or oesophagogastric junction adenocarcinoma: an open-label, dose de-escalation phase 1b study and a double-blind, randomised phase 2 study.Lancet Oncol 15: 1007-1018.
- Eunice LK, Hamid O, Filip J, Kittaneh M, Thomas Catenacci DV, et al. (2015) Clinical activity of AMG 337, an oral MET kinase inhibitor, in adult patients (pts) with MET-amplified gastroesophageal junction (GEJ), gastric (G), or esophageal (E) cancer, in GI ASCO : San Francisco.
- Kong-Beltran M,Stamos J, Wickramasinghe D (2004) The Sema domain of Met is necessary for receptor dimerization and activation.Cancer Cell 6: 75-84.
- Pennacchietti S,Cazzanti M, Bertotti A, Rideout WM 3rd, Han M, et al. (2014) Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs.Cancer Res 74: 6598-6609.
- Khoury H,Naujokas MA, Zuo D, Sangwan V, Frigault MM, et al. (2005) HGF converts ErbB2/Neu epithelial morphogenesis to cell invasion.MolBiol Cell 16: 550-561.
- Liu Y,Chattopadhyay N, Qin S, Szekeres C, Vasylyeva T, et al. (2009) Coordinate integrin and c-Met signaling regulate Wnt gene expression during epithelial morphogenesis.Development 136: 843-853.
- Koraishy FM, Silva C, Mason S, Wu D, Cantley LG (2014) Hepatocyte growth factor (Hgf) stimulates low density lipoprotein receptor-related protein (Lrp) 5/6 phosphorylation and promotes canonical Wnt signaling.J BiolChem 289: 14341-14350.
- Boon EM, van der Neut R, van de Wetering M, Clevers H, Pals ST (2002) Wnt signaling regulates expression of the receptor tyrosine kinase met in colorectal cancer.Cancer Res 62: 5126-5128.
- Monga SP, Mars WM, Pediaditakis P, Bell A, Mulé K, et al. (2002) Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes.Cancer Res 62: 2064-2071.
- Peters S,Adjei AA (2012) MET: a promising anticancer therapeutic target.Nat Rev ClinOncol 9: 314-326.
- Liu L,Zeng W, Wortinger MA, Yan SB, Cornwell P, et al. (2014) LY2875358, a neutralizing and internalizing anti-MET bivalent antibody, inhibits HGF-dependent and HGF-independent MET activation and tumor growth.Clin Cancer Res 20: 6059-6070.
- Sreekala M, B.S.R, Lida K, Gerald McM, Yaron H, et al. (2015) Blocking activity of a novel anti-MET humanized monoclonal antibody, KTN0216, is enhanced by IgG2 isotype in HGF-dependent and Met-amplified tumors in AACR Annual Meeting : Philadelphia.
- Hu S, Fu W, Xu W, Yang Y, Cruz M, et al. (2015) Four-in-one antibodies have superior cancer inhibitory activity against EGFR, HER2, HER3, and VEGF through disruption of HER/MET crosstalk.Cancer Res 75: 159-170.

