Keywords
Inflammatory reflex arc; Inflammation; Cytokines; Neuroscience; Immunology
Introduction
The inflammatory response is characterized by multifaceted interactions between pro-inflammatory and anti-inflammatory cytokines directed at eliminating pathogens, promoting healing, and reestablishing organismal homeostasis. Recently, neural reflex circuits have been demonstrated to exhibit some control of pro-inflammatory concentration, and, thus, control of the inflammatory response [1]. Known as the inflammatory reflex arc, this molecular pathway derives from vagal afferent and efferent nerve fibers [1]. The afferent arc is dependent upon vagal receptors, which sense specific ligands indicating tissue injury [1]. Once activated, the afferent arc will stimulate the efferent arc, the cholinergic anti-inflammatory pathway [1]. The cholinergic anti-inflammatory pathway regulates immunologically-mediated inflammation via suppression of pro-inflammatory cytokine expression [1]. Recent research has demonstrated that this pathway can be modulated with vagus nerve stimulation, providing a potential therapeutic option for a variety of inflammatory conditions [1]. Here, we review the molecular immunological mechanisms of the inflammatory reflex arc. Moreover, we examine the current basic science literature and consider potential clinical significance of the cholinergic anti-inflammatory pathway.
Neuroanatomy of the Vagus Nerve
Derived from the Latin vagary, meaning “wandering,” the vagus nerve acquired such a name due to its extensive distribution throughout homo sapien architecture [2]. Cranial nerve X, also known as the pneumogastric nerve, forms via a series of nerve rootlets from the lateral portions of the medulla oblongata which exit the cranium through the jugular foramen amongst the glossopharyngeal and spinal accessory nerves (Figure 1A) [2]. Vagus has a superior ganglion, the jugular ganglion, which permits general sensation and an inferior ganglion, the nodose ganglion, which allows for visceral and special sensation [2,3]. The vagus nerve has four nuclei within the medulla oblongata (Figure 1B). Primary terminal nuclei of vagal afferents include the spinal nucleus of the trigeminal nerve, receiving general somatic afferent terminations, and the nucleus tractus solitarius, collecting general visceral afferent and special visceral afferent terminations [2,3]. Nuclei of vagal efferents include the nucleus ambiguus, giving rise to somatic visceral efferents and general visceral efferents, and the dorsal motor nucleus, giving rise to general visceral efferents [2,3]. Vagus supplies motor innervation to the glands and involuntary musculature of the: tracheobronchial tree, esophagus, heart, and the alimentary tract as far as the splenic flexure [2]. Vagus also supplies motor innervation to the voluntary musculature of the superior esophagus and larynx and sensory innervation to the larynx and pharynx [2,3]. Such vast innervation permits the brain to constantly analyze and influence the physiologic status of peripheral tissue. Vagus is able to respond to a plethora of environmental stimuli via numerous receptors such as those for stretch, mechanical pressure, osmotic pressure, temperature change, and pain [4,5]. Furthermore, vagal afferents can be stimulated in response to chemical stimuli such as glucagon-like peptide-1, cholecystokinin, somatostatin, serotonin, and importantly, interleukin-1 (IL-1) [4,5].
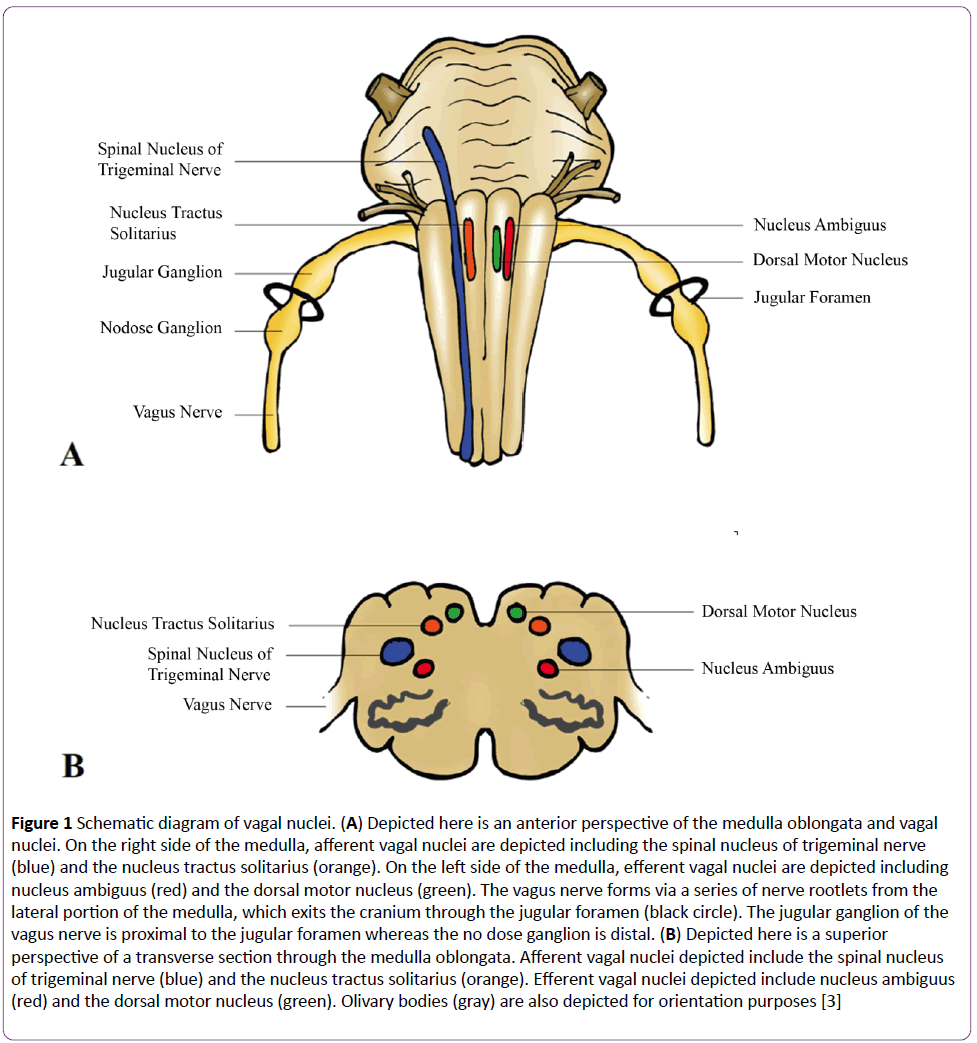
Figure 1: Schematic diagram of vagal nuclei. (A) Depicted here is an anterior perspective of the medulla oblongata and vagal nuclei. On the right side of the medulla, afferent vagal nuclei are depicted including the spinal nucleus of trigeminal nerve (blue) and the nucleus tractus solitarius (orange). On the left side of the medulla, efferent vagal nuclei are depicted including nucleus ambiguus (red) and the dorsal motor nucleus (green). The vagus nerve forms via a series of nerve rootlets from the lateral portion of the medulla, which exits the cranium through the jugular foramen (black circle). The jugular ganglion of the vagus nerve is proximal to the jugular foramen whereas the no dose ganglion is distal. (B) Depicted here is a superior perspective of a transverse section through the medulla oblongata. Afferent vagal nuclei depicted include the spinal nucleus of trigeminal nerve (blue) and the nucleus tractus solitarius (orange). Efferent vagal nuclei depicted include nucleus ambiguus (red) and the dorsal motor nucleus (green). Olivary bodies (gray) are also depicted for orientation purposes [3]
The Inflammatory Reflex Arc: The Afferent Arm
The inflammatory reflex can be activated centrally or peripherally [1,6]. Central activation is mediated by high concentrations of immunologic stimuli, such as tumor necrosis factor (TNF), acquiring access to circumventricular regions of the brain [1,6,7]. In fact, the dorsal vagal complex, which includes the dorsal motor nucleus, the area postrema, and the sensory nuclei of the nucleus tractus solitarius, has been demonstrated to exhibit a response to circulating TNF thereby modulating vagal efferent activity [6-10]. Relative to the high threshold of activation of central receptors, peripheral activation can be achieved with immunologic substrates at much lower concentrations [6,7]. Peripheral activation of vagal afferents exhibit an imperative role when the localized concentration of inflammatory mediators is not sufficient to reach the brain parenchyma from the blood [11]. The molecular mechanisms by which vagal afferents “sense” the propinquity of inflammatory mediators have yet to be fully depicted; however, vagal nerve fibers have been shown to express IL-1 receptor [5,6,11].
Previous studies have confirmed the necessity of vagal modulatory activity with respect to peripheral activation [12-16]. Watkins et al. demonstrated that the inception of fever is dependent upon vagal fibers transmitting signals from the abdomen to the brain [15]. Subdiaphragmatic vagatomy prevented the induction of fever in animals given IL-1 whereas IL-1 given to animals with an intact vagus nerve exhibited fever onset [15]. It was concluded that the vagus nerve is capable of detecting IL-1 in the periphery, transmitting this molecular information to the hypothalamus, and, as a result, increasing the activity of these neural networks associated with the onset of fever [15]. Furthermore, additional evidence has demonstrated that IL-1 increases the firing rate of vagal afferent fibers and that IL-1 stimulates neuronal depolarization [17,18].
Although IL-1 exhibits a significant role in stimulation of the afferent arc of the inflammatory reflex, other exogenous and endogenous ligands may indirectly stimulate this pathway via upregulation of IL-1 [19-21]. Exogenous ligands, such as the pathogen-associated molecular patterns (PAMPs) lipopolysaccharide and double-stranded RNA, interact with Toll-like receptors (TLRs) [19,20]. Interaction of a PAMP with a TLR initiates downstream signaling, which activates the regulatory transcription factor nuclear factor – κB (NF-κB) leading to upregulation of pro-inflammatory cytokines, including IL-1 [19,20]. Endogenous ligands, such as uric acid, heat shock proteins or other damage-associated molecular patterns (DAMPs) can also activate TLRs and produce a similar response [19,21]. The presence of PAMPs or DAMPs causing an upregulation in IL-1 levels is an additional indirect mechanism by which the afferent arm can be activated [19-21].
The Inflammatory Reflex Arc: The Efferent Arm
The afferent pathway is a fundamental component of the inflammatory reflex arc [1]. Vagal afferent fibers send signals to the brain stem, which are transduced to the nucleus tractus solitarius [3]. Polysynaptic signals connect to the autonomic nervous system’s output centers, the vagal motor neurons and the rostral ventrolateral medullary sympathoexcitatory neurons of the nucleus ambiguus, and the dorsal motor nucleus [3]. Outflow from the medulla projects to the celiac ganglion via vagal efferent fibers or preganglionic efferent nerves, which originate within the sympathetic trunk [3]. Vagal efferents terminate within the celiac ganglion [1-3]. Originating within the celiac ganglion are nerve cell bodies, the axons of which constitute the splenic nerve [22-26]. Vagal nerve fibers develop synapses on these cell bodies within the celiac ganglion, which permits control of the innate immune response in the spleen [3,22-26]. Here, the vagus nerve activates catecholaminergic splenic nerves to release norepinephrine [23,24]. Circulating norepinephrine within the splenic parenchyma interacts with beta-2-adrenergic receptor (ß2AR)-expressing T cells (CD4+Foxp3+CD44hiCD62Llo) (Figure 2) [23,27]. Adrenergic stimulation of the ß2AR induces these T cells, expressing choline acetyltransferase, to synthesize and release acetylcholine within splenic tissue [23,27]. Acetylcholine binds to splenic macrophages through the alpha7 subunit nicotinic acetylcholine receptor (α7nAChR), which is coupled to Janus kinase 2 (JAK2) and signal transducer and activator of transcription 3 (STAT3) signal transduction pathway (Figure 3) [28-30]. With respect to the splenic macrophage, binding of acetylcholine to the α7nAChR activates JAK2, a cytokine-associated tyrosine kinase receptor [28-30]. Activated JAK2 phosphorylates monomeric STAT3 promoting homo- or hetero-dimerization and translocation to the nucleus [28-30]. STAT3 interacts with NF-κB within the nucleus via protein-protein interactions thereby suppressing the transcriptional activity of NF-κB [28-30]. Furthermore, NF- κB inhibition may be achieved independent of the JAK2/STAT3 pathway [16,30]. Although not entirely defined, this second mechanism is thought to be dependent upon α7nAChR inhibiting the phosphorylation of inhibitor of NF-κB (IκB) [30]. Dephosphorylated IκB is active and remains bound to NF-κB thereby inhibiting NF-κB from translocating to the nucleus and activating gene transcription [30]. Thus, the JAK2/STAT3 and IκB pathways both suppress the transcription of various proinflammatory cytokine genes, such as IL-1, IL-6, IL-8, TNF, and CRP due to vagal efferent stimulation [22-30]. Moreover, the production of anti-inflammatory cytokines, such as transforming growth factor-ß (TGF-ß) and IL-10, is not suppressed with stimulation of the cholinergic antiinflammatory pathway; rather, concentrations of antiinflammatory cytokines remain constant [31-33].
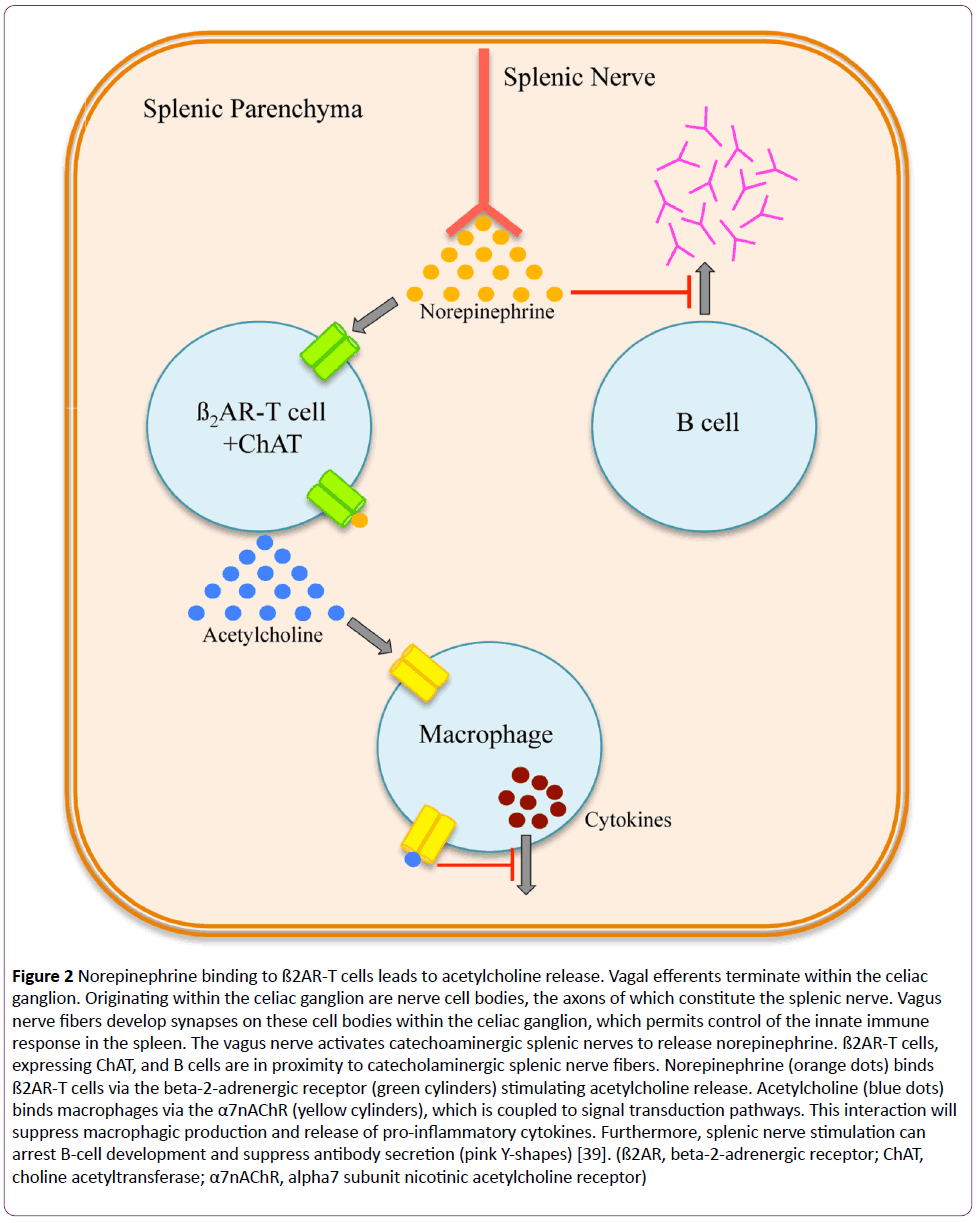
Figure 2: Norepinephrine binding to ß2AR-T cells leads to acetylcholine release. Vagal efferents terminate within the celiac ganglion. Originating within the celiac ganglion are nerve cell bodies, the axons of which constitute the splenic nerve. Vagus nerve fibers develop synapses on these cell bodies within the celiac ganglion, which permits control of the innate immune response in the spleen. The vagus nerve activates catechoaminergic splenic nerves to release norepinephrine. ß2AR-T cells, expressing ChAT, and B cells are in proximity to catecholaminergic splenic nerve fibers. Norepinephrine (orange dots) binds ß2AR-T cells via the beta-2-adrenergic receptor (green cylinders) stimulating acetylcholine release. Acetylcholine (blue dots) binds macrophages via the α7nAChR (yellow cylinders), which is coupled to signal transduction pathways. This interaction will suppress macrophagic production and release of pro-inflammatory cytokines. Furthermore, splenic nerve stimulation can arrest B-cell development and suppress antibody secretion (pink Y-shapes) [39]. (ß2AR, beta-2-adrenergic receptor; ChAT, choline acetyltransferase; α7nAChR, alpha7 subunit nicotinic acetylcholine receptor)
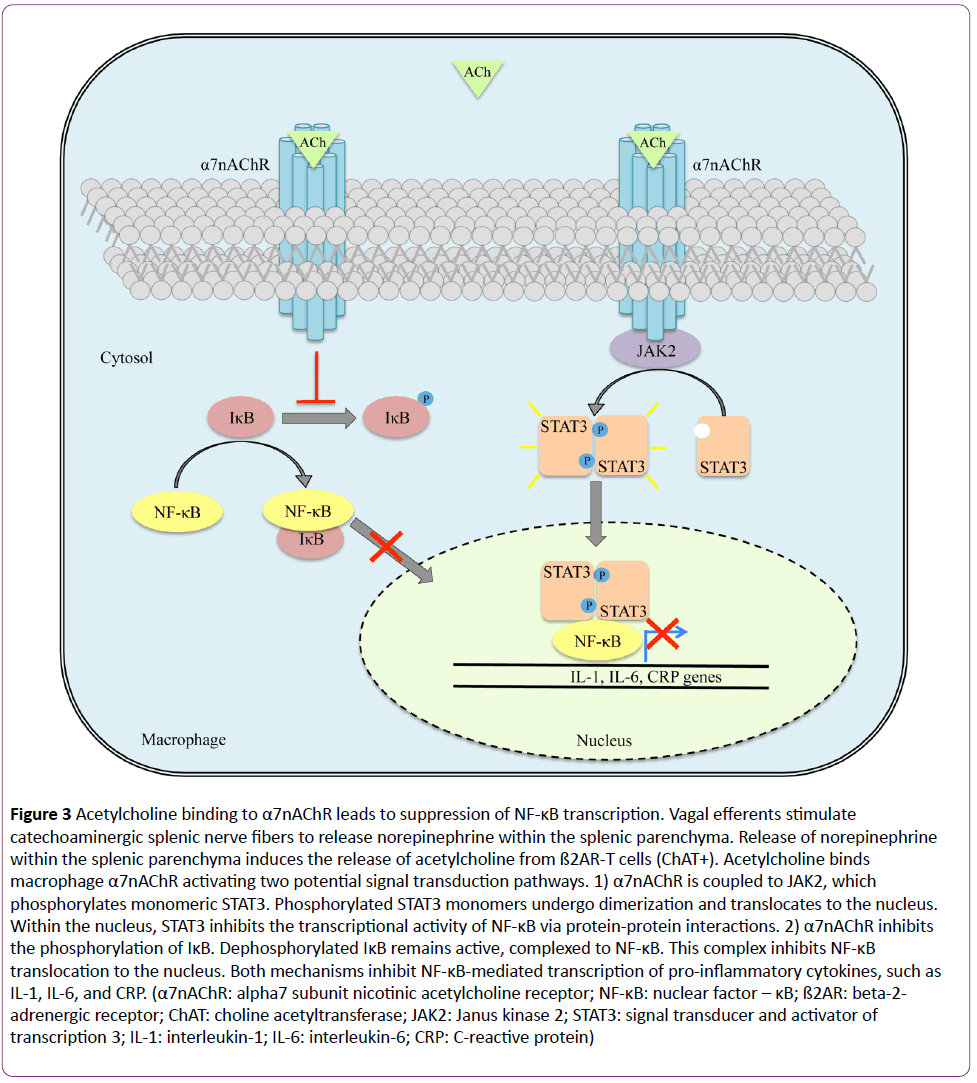
Figure 3: Acetylcholine binding to α7nAChR leads to suppression of NF-κB transcription. Vagal efferents stimulate catechoaminergic splenic nerve fibers to release norepinephrine within the splenic parenchyma. Release of norepinephrine within the splenic parenchyma induces the release of acetylcholine from ß2AR-T cells (ChAT+). Acetylcholine binds macrophage α7nAChR activating two potential signal transduction pathways. 1) α7nAChR is coupled to JAK2, which phosphorylates monomeric STAT3. Phosphorylated STAT3 monomers undergo dimerization and translocates to the nucleus. Within the nucleus, STAT3 inhibits the transcriptional activity of NF-κB via protein-protein interactions. 2) α7nAChR inhibits the phosphorylation of IκB. Dephosphorylated IκB remains active, complexed to NF-κB. This complex inhibits NF-κB translocation to the nucleus. Both mechanisms inhibit NF-κB-mediated transcription of pro-inflammatory cytokines, such as IL-1, IL-6, and CRP. (α7nAChR: alpha7 subunit nicotinic acetylcholine receptor; NF-κB: nuclear factor – κB; ß2AR: beta-2- adrenergic receptor; ChAT: choline acetyltransferase; JAK2: Janus kinase 2; STAT3: signal transducer and activator of transcription 3; IL-1: interleukin-1; IL-6: interleukin-6; CRP: C-reactive protein)
Current Research
Recently, electrical vagus nerve stimulation (VNS), activating the cholinergic anti-inflammatory pathway, has shown promising results in animal models for the treatment of a variety of inflammatory conditions [34-39]. Levine et al. considered whether VNS, via stimulation of the cholinergic anti-inflammatory pathway, can reduce disease severity in a collagen-induced arthritis (rheumatoid arthritis) animal model [34]. Animals were subject to either active or sham VNS [34]. The experimental protocol was 15 days in length [34]. On day 1, collagen-induced arthritis was induced in rats containing vagus nerve electrodes [34]. Vagus nerve stimulation was applied once daily for 60 seconds days 9 through 15 [34]. Compared to the sham group, VNS of the cholinergic antiinflammatory pathway demonstrated a significant reduction in ankle diameter and histological arthritis score [34]. The reduced histological arthritis score showed significant improvements in inflammation, cartilage destruction, pannus formation, as well as bone erosion [34]. Importantly, the improvement of bone erosion was coupled to a significant reduction in serum concentrations of receptor activator of NF- κB [34]. This study indicates that VNS, via the cholinergic antiinflammatory pathway, can significantly improve the symptomatology of collagen-induced arthritis in an animal model and may warrant future studies in human rheumatoid arthritis.
Jiang et al. investigated the role VNS may have in cerebral ischemia and reperfusion injury and the molecular mechanisms by which such a therapy may control these pathologic processes [35]. An intraluminal occlusion procedure was implemented in rats for right middle cerebral artery occlusion (MCAO) [35]. Subsequent to right MCAO, VNS was applied for 30 minutes [35]. Murine neurological function and pro-inflammatory cytokine levels were analyzed 24 hours post MCAO [35]. Rats treated with VNS exhibited significantly better neurological deficits scores as well as reduced cerebral infarct volume [35]. Furthermore, rats treated with VNS exhibited significantly decreased levels of pro-inflammatory cytokines, including IL-1ß, IL-6, and TNF- α, within the brain penumbra [35]. This study signifies that VNS is a neuroprotective therapy in acute cerebral ischemia via activation of the cholinergic anti-inflammatory pathway [35].
Rezende-Neto et al. hypothesized that coagulopathy may be improved with VNS via molecular modulation of the inflammatory pathway to hemorrhagic shock [36]. Rats were divided into three groups: sham hemorrhagic shock (HS), HS without VNS, and HS with VNS [36]. Hemorrhage was induced by withdrawing 40% of total blood volume [36]. No fluids were administered for 15 minutes with the intention of simulating the time period prior to the arrival of emergency medical services [36]. Fluid resuscitation was then implemented for 45 minutes [36]. Next, rats were subject to VNS in discrete intervals for a total of 3.5 minutes [36]. Relative to baseline, rats who were given VNS post-HS revealed a significant increase in maximum clot firmness [36]. Importantly, a significant increase in the pro-inflammatory cytokine IL-1 was observed in rats subject to HS without VNS whereas a significant decrease in IL-1 was observed in rats subject to HS with VNS [36]. Furthermore, the anti-inflammatory cytokine IL-10 was observed to be significantly elevated in rats subject to HS with VNS [36]. These results indicated that VNS in rats subject to HS improves coagulation as well as decreases the inflammatory response to hemorrhage via increased activity of the cholinergic anti-inflammatory pathway [36].
Conclusion
In this brief review, we described the inflammatory reflex arc in detail, with an emphasis placed on potential stimulation of the cholinergic anti-inflammatory pathway. Indeed, the inflammatory reflex arc has transformed our perspective on the neuroimmunology of inflammation. Potential neuroimmunomodulation of the cholinergic anti-inflammatory pathway may very well be a promising therapeutic option for inflammatory conditions in the future.
Conflicts of Interest
None to report.
9123
References
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, et al. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458-462. Retrieved from https://www.nature.com/nature/journal/v405/n6785/full/405458a0.html
- Moore KL (2013) Clinically oriented anatomy. (7thedn), Lippincott Williams & Wilkins, Philadelphia, PA.
- Babic T, Browning KN (2013) The role of vagal neurocircuits in the regulation of nausea and vomiting. Eur J Pharmacol 722: 38-47. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3893663/
- Mussa BM, Sartor DM, Verberne AJ (2008) Activation of cholecystokinin (CCK 1) and serotonin (5-HT 3) receptors increases the discharge of pancreatic vagal afferents. Eur J Pharmacol 601: 198-206. Retrieved from https://www.sciencedirect.com/science/article/pii/S0014299908011400
- Berthoud HR, Neuhuber WL (2000) Functional and chemical anatomy of the afferent vagal system. AutonNeurosci 85: 1-17. Retrieved from https://www.autonomicneuroscience.com/article/S1566-0702(00)00215-0/abstract
- Borovikova LV, Ivanova S, Nardi D, Zhang M, Yang H, et al. (2000) Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. AutonNeurosci 85: 141-147. Retrieved from https://www.autonomicneuroscience.com/article/S1566-0702(00)00233-2/abstract
- Emch GS, Hermann GE, Rogers RC (2000) TNF-alpha activates solitary nucleus neurons responsive to gastric distension. Am J PhysiolGastrointest Liver Physiol 279: G582-G586. Retrieved from https://ajpgi.physiology.org/content/279/3/G582
- Goehler LE, Gaykema RP, Hansen MK, Anderson K, Maier SF, et al. (2000) Vagal immune-to-brain communication: a visceral chemosensory pathway. AutonNeurosci 85: 49-59. Retrieved from https://www.autonomicneuroscience.com/article/S1566-0702(00)00219-8/abstract
- Hermann GE, Emch GS, Tovar CA, Rogers RC (2001) c-Fos generation in the dorsal vagal complex after system endotoxin is not dependent on the vagus nerve. Am J PhysiolRegulIntegr Comp Physiol 280: R289-R299. Retrieved from https://ajpregu.physiology.org/content/280/1/R289.article-info
- Romanovsky AA (2000) Thermoregulatory manifestations of system inflammation: lessons from vagatomy. AutonNeurosci 85: 39-48. Retrieved from https://www.autonomicneuroscience.com/article/S1566-0702(00)00218-6/abstract
- Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, et al. (1997) Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull 43: 357-364. Retrieved from https://www.sciencedirect.com/science/article/pii/S0361923097000208
- Hansen MK, Nguyen KT, Goehler LE, Gaykema RP, Fleshner M, et al. (2000) Effects of vagotomy on lipopolysaccharide-induced brain interleukin-1beta protein in rats. AutonNeurosci 85: 119–126. Retrieved from https://europepmc.org/abstract/MED/11189018
- Hansen MK, O’Connor KA, Goehler LE, Watkins LR, Maier SF (2001) The contribution of the vagus nerve in interleukin-1beta-induced fever is dependent on dose. Am J PhysiolRegulIntegr Comp Physiol 280: R929–R934. Retrieved from https://ajpregu.physiology.org/content/280/4/R929.long
- Watkins LR, Maier SF (1999) Implications of immune-to-brain communication for sickness and pain. ProcNatlAcadSci USA 96: 7710–7713. Retrieved from https://www.pnas.org/content/96/14/7710.long
- Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, et al. (1995) Blockade of interleukin-1 induced hyperthermia by subdiaphragmaticvagotomy: evidence for vagal mediation of immune-brain communication. NeurosciLett 183: 27–31. Retrieved from https://www.ingentaconnect.com/content/els/03043940/1995/00000183/00000001/art11105.
- Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, et al. (2002) Pharmacological stimulation of the cholinergic anti-inflammatory pathway. J Exp Med 195: 781-788.Retrieved from https://jem.rupress.org/content/195/6/781.long
- Niijima A (1996) The afferent discharges from sensors for interleukin 1 beta in the hepatoportal system in the anesthetized rat. J AutonNervSyst 61: 287-291. Retrieved from https://www.sciencedirect.com/science/article/pii/S0165183896000987
- Ferri CC, Yuill EA, Ferguson AV (2005) Interleukin-1beta depolarizes magnocellular neurons in the paraventricular nucleus of the hypothalamus through prostaglandin-mediated activation of a non-selective cationic conductance. RegulPept 129: 63-71. Retrieved from https://www.sciencedirect.com/science/article/pii/S0167011505000170
- Areal H, Abrantes J, Esteves PJ (2011) Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evolutionary Biology 11: 368. Retrieved from https://bmcevolbiol.biomedcentral.com/articles/10.1186/1471-2148-11-368
- Pistoia V, Raffaghello L (2011) Damage-associated molecular patterns (DAMPs) and mesenchymal stem cells: a matter of attraction and excitement. Eur J Immunol 41:1828-1831. Retrieved from https://onlinelibrary.wiley.com/doi/10.1002/eji.201141724/abstract;jsessionid=B934C32F6034E7B2DBDB5BF38DA35BDC.f01t04
- Lotfi R, Eisenbacher J, Solgi G, Fuchs K, Yildiz T, et al. (2011) Human mesenchymal stem cells (MSCs) respond to native but not oxidized damage associated molecular pattern molecules (DAMPs) from necrotic (tumor) material. Eur J Immunol 41: 2021–2028. Retrieved from https://onlinelibrary.wiley.com/doi/10.1002/eji.201041324/full
- Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, et al. (2014) Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol 7: 335-347. Retrieved from https://www.nature.com/mi/journal/v7/n2/full/mi201352a.html
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, et al. (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334: 98-101. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4548937/
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, et al. (2008) Splenic nerve is required for cholinergic anti-inflammatory pathway control of TNR in endotoxemia. ProcNatlAcadSci USA 105: 11008-11013. Retrieved from https://www.pnas.org/content/105/31/11008.full
- Huston JM, Rosas-Ballina M, Xue X, Dowling O, Ochani K, et al. (2009) Cholinergic neural signals to the spleen down-regulate leukocyte trafficking via CD11b. J Immunol 183: 552-559. Retrieved from https://www.jimmunol.org/content/183/1/552.long
- Huston JM, Ochani M, Rosas-Ballina M, Liao H, Ochani K, et al. (2006) Splenectomy inactivates the cholinergic anti-inflammatory pathway during lethal endotoxemia and polymicrobial sepsis. J Exp Med 203: 1623-1628. Retrieved from https://jem.rupress.org/content/203/7/1623.full
- Olofsson PS, Katz DA, Rosas-Ballina M, Levine YA, Ochani M, et al. (2012) α7 nicotinic acetylcholine receptor (α7nAChR) expression in bone marrow-derived non-T cells is required for the inflammatory reflex. Mol Med 18: 539-543. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3356417/
- de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, et al. (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6: 844-851. Retrieved from https://www.nature.com/ni/journal/v6/n8/full/ni1229.html
- Yu Z, Zhang W, Kone BC (2002) Signal transducers and activators of transcription 3 (STAT3) inhibits transcription of the inducible nitric oxide synthase gene by interacting with nuclear factor kappaB. Biochem J 367: 97-105. Retrieved from https://www.biochemj.org/content/367/1/97.long
- Yoshikawa H, Kurokawa M, Ozaki N, Nara K, Atou K, et al. (2006) Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. ClinExpImmunol 146: 116-123. Retrieved from https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2249.2006.03169.x/abstract
- Hilderman M, Qureshi AR, Al-Abed Y, Abtahi F, Lindecrantz K, et al. (2015) Cholinergic anti-inflammatory pathway activity in dialysis patients: a role for neuroimmunomodulation? Clin Kidney J 8: 599-605. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4581391/
- Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, et al. (2008) Modulation of TNF release by choline requires alpha7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med 14: 567–574. Retrieved from https://static.smallworldlabs.com/molmedcommunity/content/pdfstore/567_574.Parrish.00079.PDF
- Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, et al. (2007) Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med 35: 1139-1144. Retrieved from https://journals.lww.com/ccmjournal/pages/articleviewer.aspx?year=2007&issue=04000&article=00020&type=abstract
- Levine YA, Koopman FA, Faltys M, Caravaca A, Bendele A, et al. (2014) Neurostimulation of the cholinergic anti-inflammatory pathway ameliorates disease in rat collagen-induced arthritis. PLoS One 9: e104530. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4128811/
- Jiang Y, Li L, Liu B, Zhang Y, Chen Q, et al. (2014) Vagus nerve stimulation attenuates cerebral ischemia and reperfusion injury via endogenous cholinergic pathway in rat. PLoS One 9: e102342. Retrieved from https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0102342
- Rezende-Neto JB, Alves RL, CarvalhoJr M, Almeida T, Trant C, et al. (2014) Vagus nerve stimulation improves coagulopathy in hemorrhagic shock: a thromboelastometric animal model study. J Trauma Manag Outcomes 8:15. Retrieved from https://www.traumamanagement.org/content/8/1/15
- Li P, Liu H, Sun P, Wang X, Wang C, et al. (2016) Chronic vagus nerve stimulation attenuates vascular endothelial impairments and reduces the inflammatory profile via inhibition of the NF-KB signaling pathway in ovariectomized rats. ExpGerontol 74: 43-55. Retrieved from https://www.sciencedirect.com/science/article/pii/S0531556515301042
- Cheng Z, Li-Sha G, Jing-Lin Z, Wen-Wu, Xue-Si C, et al. (2014) Protective role of the cholinergic anti-inflammatory pathway in a mouse model of viral myocarditis. PLoS One 9: e112719. Retrieved from https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0112719
- Mino-Osorio P, Rosas-Ballina M, Valdes-Ferrer SI, Al-Abed Y, Tracey KJ, et al. (2012) Neural signaling in the spleen controls B-cell responses to blood-borne antigen. Mol Med 18: 618-627. Retrieved from https://static.smallworldlabs.com/molmedcommunity/content/pdfstore/12_027_Mina-Osorio.pdf

