Keywords
Lipoarabinomannan, Toll like Receptor-2, NF-kB, Cytokines, Protein Kinases/Phosphatases, Signal Transduction.
Introduction
Mycobacterium tuberculosis, the causative pathogen of the dreaded disease tuberculosis, infects and multiplies within the granulomas created in the lungs [1, 2]. Upon invasion, the bacterial lipid antigens induce secretion of chemotactic factors from mononuclear cells that cause further aggregation of T cells, B cells, monocytes and macrophages [3, 4] and gradually this inflammatory response is suppressed and the bacteria flourishes within the granulomas [5, 6].
Research regarding tuberculosis has been very actively expanding in recent years. It is known that the mycobacterial species produce a large amount of cell wall glycolipid antigen called Lipoarabinomannan (LAM), responsible for the virulence of the species [5, 6] and induces the alteration of the normal cell signaling pathways [7-9]. Virulent Mycobacterial strains possess mannose capping on the oligoarabinofuranosyl side chains at the non-reducing termini of the glycolipid, thereby yielding Mannosylated Lipoarabinomannan (Man-LAM), whereas the avirulent strains (Mycobacterium smegmatis, Mycobacterium tuberculosis H37 Ra) are devoid of these mannose capping, hence yielding Arabinosylated Lipoarabinomannan (Ara-LAM) [10, 11]. Man-LAM induces the survival of the infected cell by increasing the expression of cell survival factors like Phosphoinositol 3’ kinase (PI3K), Protein Kinase B [Akt] [12, 13] and MAPK/ERK [14], while Ara-LAM treatment of host PBMC has been reported to facilitate cellular cytotoxicity and apoptosis [15].
However, the role of cytokines (low molecular weight soluble effectors of cell functions) is of great importance in maintaining the normal functions of a cell. Pro-inflammatory cytokines like TNF-α, IL-12 and IFN-γ secretion are reported to be up regulated by Ara-LAM treatment of macrophages, but not by Man-LAM treatment [16-18], resulting in a subsequent increase in iNOS2 mRNA expression and nitrite generation [19]. Ara-LAM is also reported to utilize Toll-like receptors (TLR), a family of receptors that are responsible for activation of macrophages by Mycobacterial products [20]. Inflammatory processes within the lung as well as pulmonary macrophages are induced by Ara-LAM via TLR-2 [21], which are essential for the induction of pro-inflammatory cytokines [22]. On the other hand, treatment of macrophages with Mycobacterium tuberculosis lipoarabinomannan induces IRAK-M and negatively regulates Toll-like receptor-dependent Interleukin-12 p40 production in macrophages [23]; besides, it potently induces the release of anti-inflammatory (Th2) cytokines such as Interleukin-10 (IL-10) and Transforming Growth factor beta (TGF-β) [24]. IL-10 is immunosuppressive in nature thus helping in the establishment of pathogenesis [25, 26]. The ratio between pro- and anti-inflammatory cytokines is a very important parameter determining cell function as depicted by the TNF-α: IL-10 ratio; the lower this ratio, the greater is the chance of developing pathogenesis due to the survival of the infected cell [27]. Not only that, recent reports from our laboratory have shown that Ara-LAM has immunomodulatory role in Leishmania donovani infected murine macrophages [28], and this molecule also induces protection against visceral leishmaniasis through up-regulation of TLR-2 signaling [29].
Earlier anti-tuberculosis researches have been able to deliver some primary line antibiotics like Isoniazid (INH), Pyrazinamide, Ethambutol and Rifampicin but unfortunately the pathogen has acquired resistance mutations against these drugs. Hence, newer approaches aimed at targeting the immune mechanisms of the host need to be devised as alternative medication. Recent studies have shown mycobacterial lipid antigens and oligosaccharide protein conjugates are highly immunogenic against the virulent pathogen [30, 31].
In this paper, we have found that Ara-LAM could potently confer protection against the cell signaling alterations caused by the virulent Man-LAM in murine model, both in vitro and in vivo. In both cases we observed a strong induction of proinflammatory cytokines TNF-α, IL-12 and IFN-γ and subsequently nitric oxide generation under Ara-LAM treated condition. A further look into the upstream mechanisms established that this protective effect was mediated by increased NF-kB binding to DNA due to TLR-2 induction by Ara-LAM. These findings could lead to a novel approach targeting cell signaling mediated immunotherapy against mycobacterial diseases.
Materials and Methods
Materials
Anti TLR-2 T2.5 murine clone mAb (Hycult Biotech, Uden, The Netherlands), rabbit anti MyD88, rabbit anti IRAK-1 and rabbit anti Ikk-α/β antibodies, as well as TLR-2-PE conjugated primary antibody were purchased from Santa Cruz Biotech, Santa Cruz, CA, USA. Oligos, for semi-quantitative and Real Time PCR were purchased from Bangalore Genei, India, while TLR-2 specific siRNA from Ambion Inc, Austin, TX, USA. Oligofectamine, the transfecting agent was obtained from Invitrogen life technologies, USA. Tissue culture reagents were purchased from Gibco Laboratories, USA. TRIZOL (Sigma, USA), dNTPs, RevertAidTM M-MuLV Reverse Transcriptase, oligo dT and Taq polymerase and other chemicals required for RT-PCR were purchased from Fermentas. All other chemicals were purchased from either Sigma (USA) or MERCK. [γ-32P] ATP from JONAKI, DAE, India. SYBR Green Jump-Start Taq Ready Mix for Quantitative PCR from Sigma (USA).
Animals
C57BL/6 mice purchased from National Center for Laboratory Animal Sciences (NCLAS), India. For each experiment with Man LAM, 8-10 mice (about 6-8 weeks old) were used regardless of sex.
Isolation and Purification of LAM
Mannosylated Lipoarabinomannan (Man-LAM) from virulent Mycobacterium tuberculosis H37Rv was obtained as a kind gift from Dr. Sujoy K. Dasgupta, department of Microbiology, Bose Institute, who originally got it from Dr. John Belisle through “TB Research Materials and Vaccine Testing”, NIH, NIAID, Contract NO1-AI-75320, Colorado State University, Fort Collins, CO, USA. The endotoxin content of this product was <10 ng/μg as detected by Limulus test.
Isolation of Ara-LAM: Ara-LAM was purified from strain LR222 of M. smegmatis. All the experiments were performed with a single isolate of each culture. LAM was isolated and purified according to process as described in our previous paper [32].
Isolation of murine peritoneal macrophages
Mouse macrophages were isolated by peritoneal lavage with ice-cold PBS 48 h after intraperitoneal injection of 1.0 µl of sterile 4% thioglycolate broth (Difco). Cells were cultured as described by Fahey et al. [33]. The adherent cell population was cultured for 48 h prior to any treatment, to achieve the resting state.
LAM Treatment and siRNA transfection of cells
Preparation of TLR-2 specific siRNA:
We designed and synthesized TLR-2 siRNA (sequence mentioned in the oligos chart) using silencer siRNA construction kit (Ambion Inc.). A nonspecific scrambled siRNA were generated with same GC content.
Treatment of macrophages and transfection with siRNA
The peritoneal macrophages isolated from C57BL/6 mice were treated with Man-LAM at a specific dose of 10 μg/ µl. The Ara LAM pretreated cells (macrophages pre-incubated with Ara-LAM dose at a standardized non-lethal, non-cytotoxic dose of 3 μg/ µl for 3 hours) were followed by Man LAM treatment with the above specified dose. For siRNA transfection, the isolated macrophages were cultured in 35 mm plates (3.5x106) for 48 hours in RPMI-1640 medium supplemented with 10% FBS. The siRNA was transfected directly into murine peritoneal macrophages at a final concentration of 100 nM, using oligofectamine (Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s instructions. The macrophages were incubated with the transfection complexes in low (2%) serum for 24 hours [34], after which further treatments were done and the cells were cultured for the specified time period.
Association of signaling molecules
Co-immunoprecipitation of TLR-2/MyD88 and MyD88/IRAK-1 The adherent cell population was scraped and centrifuged at 400 x g for 15 min at 4 °C. The cells were then resuspended in ice-cold extraction buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM NaF, 1 mM EDTA, 1 mM EGTA, 1mM Na3VO4, 0.2% Nonidet P-40, 10ug/ µl aprotinin. The cell lysates were centrifuged at 14000 x g for 10 min at 4 °C. The supernatants (200 ug of protein) from the cell lylysates were subjected to immunoprecipitation with anti-TLR-2 (or anti-IRAK-1) antibody for overnight at 4 °C. The immuno complexes were captured by incubation with anti rabbit IgG agarose (Gibco-BRL) secondary antibody for 2 hours at 4 °C. The immuno complexes were then washed twice with wash buffer and twice with lysis buffer and then assessed for coimmunoprecipitation with MyD88 probing the blot with the anti-MyD88 primary antibody. Equal loading of samples was verified by reprobing the blots with anti-TLR-2. The process of western blot was performed as per earlier reports [35, 36].
IRAK-1 and Ikk kinase assays
5x106 cells were collected, pelleted at 1000xg for 10 minutes, and lysed on ice for 10 minutes in lysis buffer comprising 50 mM HEPES pH 7.6, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM benzamidine, 5 mM paranitrophenyl phosphate (PNPP), 1mM DTT, 1 mM PMSF, 1 μg/ µl each of aprotinin, leupeptin and pepstatin. The protein was estimated from the supernatant obtained by centrifugation by Bio-Rad protein estimation kit. Extracts containing equal amount of protein were used for immunoprecipitation.
5 μl of anti rabbit IRAK-1 antibody (Santa Cruz Biotech) were added to the supernatant and incubated for 3 hours at 4 ºC. 50 μl of protein A agarose beads (anti rabbit) were added to the mixture and followed by incubation for 2 hours at 4 ºC. Samples were repeatedly washed with lysis buffer, and then washed with kinase buffer comprising 20 mM HEPES pH 7.6, 20 mM MgCl2, 20 mM β-glycerophosphate, 20 mM paranitrophenylphosphate (PNPP), 1 mM EDTA, 1 mM sodium orthovanadate, 1 mM benzamidine. 50 μl of kinase buffer were added to each sample, supplemented with 5 μM ATP, 1 µg myelin basic protein (MBP), 1 μl [g-32-P] ATP, and incubated for 37 ºC for 30 minutes. SDS sample buffer was added, and the samples were subjected to SDS-PAGE analysis. The gel was dried and exposed to an X-ray film [37].
Ikk kinase assay was performed in a similar manner but the buffers used were different. The immunprecipitation buffer comprised of 50mM Tris pH 7.4, 400mM NaCl, 3 mM EDTA, 3 mM EGTA, 1% Triton X-100, 0.5% NP-40, 10% Glycerol, 1 mM PMSF, 2 mM DTT, 0.1% sodium orthovanadate, 20 mM β-glycerophosphate. The kinase buffer comprised of 50 mM/L Tris pH 7.5, 10 mM MgCl2, 20 mM β-glycerophosphate, 100 μM sodium orthovanadate, 1μM DTT, 20 μM cold ATP, 1 μg Ikk substrate protein and 1 μl [γ-32-P] ATP.
Electrophoresis and iμMunoblotting
Proteins were quantified with Bio-Rad protein assay reagent using BSA as a standard. Equal amount of protein in each lane were subjected to Sodium Dodecyl Sulfate-10% polyacrylamide gel electrophoresis (SDS-10%PAGE) and transferred to a nitrocellulose membrane. The membrane was blocked overnight with 3% bovine serum albumin in Tris-saline buffer (pH 7.5) and iμMunoblotting was done as described by Majumdar and coworkers [36].
Electrophoretic Mobility Shift Assay (EMSA)
NF-kB specific oligonucleotide (5’-TAGTTGAGGGCACTTTC CCAGG-3’) from the NF-kB/RelA DNA-binding domain in murine IkB light chain gene enhancer probe (synthesized from Gibco-BRL) were labeled with 32P with Klenow by using [α-32P] dATP. Nuclear extracts (15 µg per sample) were incubated with 3 X 105 cpm of 32P-labeled probe (0.2 ng of DNA) in the presence of binding buffer (containing 12.5 μM HEPES [pH 7.9], 10% glycerol, 5 μM MgCl2, 50 μM KCl, 1 μM DTT, and 300 µg of Bovine Serum Albumin/ µl) and 44 μg of salmon sperm DNA for 20 min. For cold competition, 10 -and 100- fold excess unlabeled probe was incubated for 15 min at room temperature with the mixture described above before the addition of the labeled probe. Shift complexes were resolved in 6% acrylamide gels at 40C in 0.5 X Trisborate- EDTA; dried gels were autoradiographed [37].
Flow Cytometric Analysis
FACS (Flourescence Activated Cell Sorter) analysis of cell surface expression of TLR-2-FITC was done. Briefly, 1x106 cells per set, after treatment, were washed with chilled PBS, collected by repeated flushing and pelleted down. They were then fixed with 1% paraformaldehyde, washed twice with wash buffer, and incubated for 1 h in dark (at 4 °C) with anti- TLR-2 conjugated with PE under shaking condition. A separate set of cells were permeabilized with 0.1% saponin and treated as explained. After the period of incubation was over, the cells were washed twice with FACS buffer (1X). Finally the cell suspension in the buffer was subjected to analysis of the membrane bound form of TLR-2 by Flow Cytometry (FACSCalibur; Recton Dickinson). Normal rabbit IgG (1:500) was used instead of primary antibody as negative control.
Cytokine assay by Sandwich ELISA
The isolated macrophages from C57BL/6 mice were treated as mentioned above. After the indicated time point, the cellfree supernatants were collected and subjected to sandwich ELISA. Sandwich ELISA kits were obtained from Factor Test-X, R&D systems. The ELISA was performed as per the instructions of the manufacturer. The detection limits of the kits were 15 pg/ µl and < 4 pg/ µl for TNF-α and IL-12 p40 respectively.
Quantification of cytokine mRNA
Isolation of RNA and RT-PCR
RNA was isolated according to the standard protocol [38, 39]. Briefly total RNA extracted from 4 x 106 macrophages using TRIZOLTM reagent (SIGMA). Isolated total RNA was then reverse transcribed using Revert AidTM M-MuLV Reverse Transcriptase (Fermentas). The cDNA encoding the gene was amplified using specific primers, for IL-12 (forward 5’-CAACATCAAGAGCAGTAGCAG- 3’; reverse 5’-TACTCCCAGCTGA CCTCCAC-3’, product size 799 bp); for TNF-α, (forward 5’-GGCAGGTCTACTTTGG AGTCATTGC-3’; reverse 5’-ACATTCGAGGCTCCAGTGAATTCGG-3’, product size 300 bp), for IFN-g (forward 5’-GGATATCTGGAGGAACTGGC-3’; reverse 5’-CGACTCCTTTTTCCGCTTCCT-3’, product size 433 bp) and for iNOS2 (forward 5’CCCTTCCGAAGTTTCTGGCAGCAGC- 3’; reverse 5’-GGCTGTCAGAGCCTCGTG GCTTTGG-3’, product size 496 bp). PCR amplification was conducted in a reaction volume of 50 µl using a Perkin Elmer Gen Amp PCR system 2400 and 0.5 unit of Taq polymerase set for 35 cycles (denaturation: at 94 ºC for 30 sec; annealing: at 58 ºC for 30 sec; extension: at 72 ºC for 30 sec) for IL-12p40 and IFNγ. For TNF-α and iNOS2, PCR reactions at 94 ºC for 30 sec, at 58 ºC 30 sec and 72 ºC for 1 min were carried out for 30 cycles. For Glyceraldehydes–3 Phosphate Dehydrogenase (GAPDH) as control was PCR amplified using, 5’-CAAGGCTGTGGGCAAGGTCA- 3’ and 5’-AGGTGGAAGAGTG GGAGTTGCTG- 3’ oligos located in different exons as sense and antisense primers, respectively. The expected size of PCR product is 242 bp. PCR amplified product was subsequently size fractioned on 1% agarose gel, stained with Ethidium Bromide and visualized under UV-light. In negative control experiments with omission of the reverse transcriptase, no PCR product was detected for either set of the PKC and GAPDH primers (data not shown).
Real Time quantitative PCR
Relative quantitative RT-PCR was performed on iCycler (Bio- Rad Laboratories, Hercules,CA) using SYBR green reagent. The PCR mixture (25 µl) contained 1.0 µl of each primer, 8 µl of water, 12.5 µl of a commercial SYBR Green PCR master mixture (SIGMA) and 2.0 µl of cDNA. The samples were placed in 96-well plates (Bio-Rad) that were sealed with optical sealing tape (Bio-Rad). PCR amplifications were performed by using the iCYCLER iQ Multi-color real-time PCR detection system (Bio-Rad). The thermal cycling conditions were 5 min at 95 ºC and then 40 cycles of 30 sec at 94 ºC, then 30 sec at 58 ºC, and then 1 min at 72 ºC followed by melt curve analysis. An internal control GAPDH was amplified in separate tubes. We have used the Comparative cycle threshold method (ΔΔCt method) for relative quantitation of gene expression. First, the Ct for the target amplicon and the Ct for the internal control (GAPDH) were determined for each sample. The ΔCt for each experimental sample was subtracted from the calibrator. This difference was called the ΔΔCt value. Finally, the arithmetic calibrator (2-ΔΔCt) was used to calculate the amount of target normalized to the amount of an internal control and relative to the amount of the calibrator. Thus, all the values for experimental samples were expressed as fold differences between the sample mRNA and the calibrator mRNA.
Nitrite generation assay
Nitrite accumulation in culture was measured colorimetrically by the Griess Reaction [40, 41] using the NO colorimetric Assay kit (Boehringer Mannheim). For each assay, macrophages were cultured in 24 well tissue culture plate (Tarson) at a concentration of 1x106 cells /ml. Cell free supernatants was collected, and the nitrite level estimated as per the instructions of the manufacturer. Data are expressed as micromoles of nitrite.
Quantitation of data
Densitometric Analysis
The RT-PCR results showing cytokine mRNA expression and the autoradiograph were analyzed using UV-Gel Camera and Molecular Analyst version 1.5software (Bio-Rad Laboratories, Hercules, Calif.)
Statistical analysis
Results were expressed as the mean plus or minus the Standard Deviation (SD) for individual sets of experiments. Each experiment was performed at least thrice, and representative data from each set were presented in the manuscript. One or two tailed Students t test for significance was performed as applicable in each case. A P value of < 0.05 was considered significant.
Results
Ara-LAM pre-treatment facilitates TLR-2 activation in Man-LAM treated macrophages
TLRs are known to play a key role in innate immune recognition and subsequent activation of adaptive immunity [22]. It is well known that inflammatory processes within the lung as well as pulmonary macrophages are induced by Ara-LAM via TLR-2 [21], essential for the induction of pro-inflammatory cytokines [22, 42]. However, Man-LAM failed to elicit pro-inflammatory cytokine gene transcription through a TLR-2 dependent mechanism. Firstly, we standardized the non-lethal dose of the cytotoxic Ara-LAM on macrophage viability to be 3µg/ml as reported in previous reports from our laboratory [28, 29]. We further investigated the effect of Ara-LAM pretreatment on the frequency of TLR-2-PE positive cells by Flow Cytometry in Man-LAM treated cells. We observed that TLR-2 was expressed at a low level constitutively in either untreated (Fig. 1a) or Man-LAM treated cells (Fig. 1b) but Ara-LAM pretreatment of Man-LAM treated cells showed a significant increase in the frequency of TLR-2-PE positive macrophages (Fig. 1d) compared to either control or Man-LAM treated macrophages. Fig. 1e shows a bar diagram expressing the percentage of TLR-2-PE positive cells in cells treated with Man-LAM, Ara-LAM and Ara-LAM pre-treated followed by Man-LAM treatment.
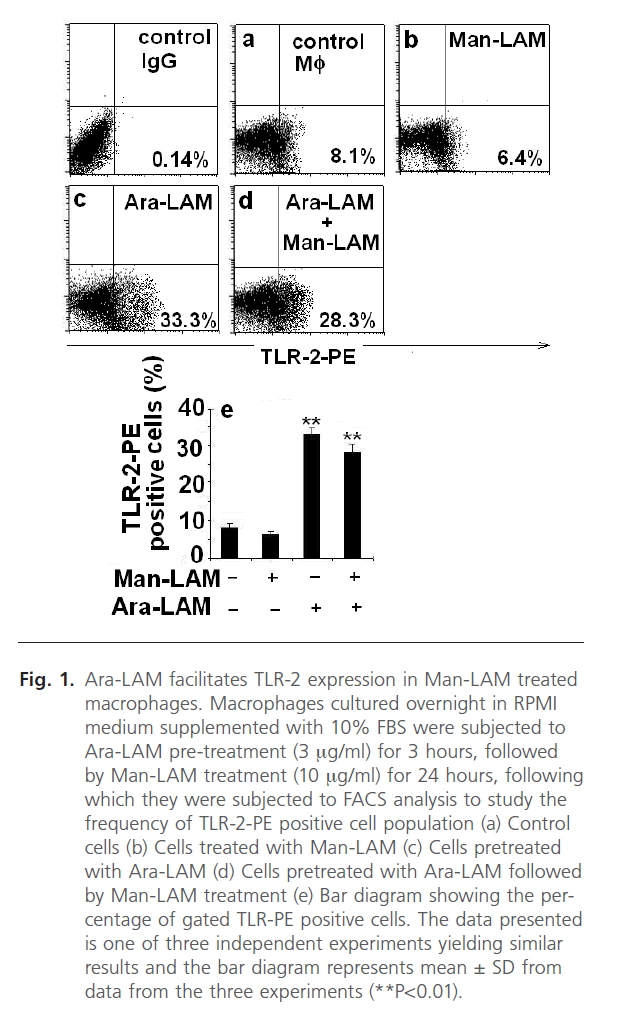
Fig 1: Ara-LAM facilitates TLR-2 expression in Man-LAM treated macrophages. Macrophages cultured overnight in RPMI medium supplemented with 10% FBS were subjected to Ara-LAM pre-treatment (3 mg/ml) for 3 hours, followed by Man-LAM treatment (10 mg/ml) for 24 hours, following which they were subjected to FACS analysis to study the frequency of TLR-2-PE positive cell population (a) Control cells (b) Cells treated with Man-LAM (c) Cells pretreated with Ara-LAM (d) Cells pretreated with Ara-LAM followed by Man-LAM treatment (e) Bar diagram showing the percentage of gated TLR-PE positive cells. The data presented is one of three independent experiments yielding similar results and the bar diagram represents mean ± SD from data from the three experiments (**P< 0.01).
Ara-LAM facilitates TLR-2-MyD88 association in Man-LAM treated macrophages
It is known that TLR-2 activation involves the association of TLR-2 with MyD88 (Myeloid Differentiation Factor 88), an adaptor molecule located immediate downstream of TLR-2 [43], and this event is crucial for the initiation of the further signals. Hence, finding that Ara-LAM pretreatment in Man- LAM treated macrophages enhanced the frequency of TLR- 2-FITC positive cells on the cell surface, we studied whether Ara-LAM induced the association of TLR-2 and MyD88 under similar conditions. For this, the cell lysates were subjected to immunoprecipitation with anti-TLR-2 antibody or control mouse IgG, and the blots were probed with anti-MyD88 antibody. A strong selective association between TLR-2 and MyD88 was observed in Man-LAM treated or untreated macrophages under Ara-LAM pre-treated condition (Fig. 2, lanes 2 and 4), compared to Man-LAM treated macrophages (Fig. 2, lane 3).
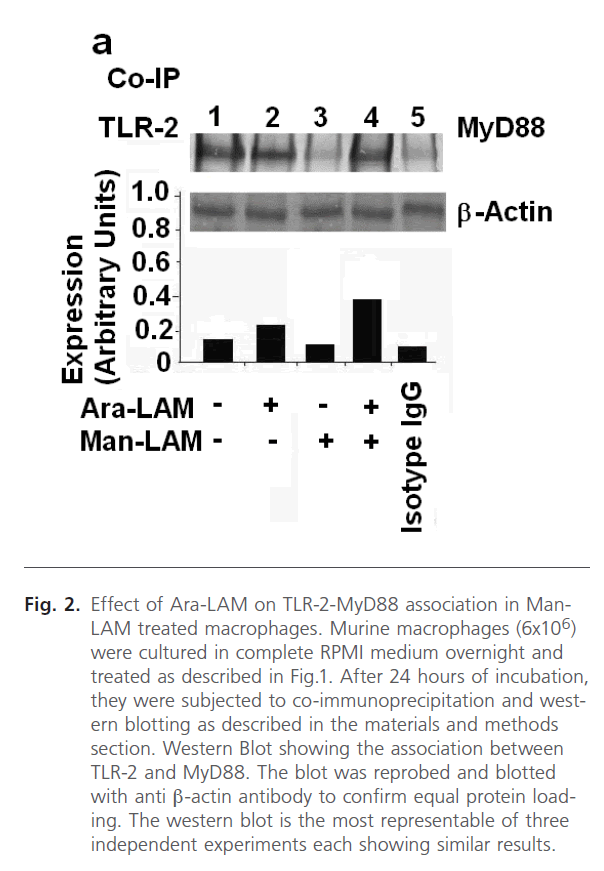
Fig 2: Effect of Ara-LAM on TLR-2-MyD88 association in Man- LAM treated macrophages. Murine macrophages (6x106) were cultured in complete RPMI medium overnight and treated as described in Fig.1. After 24 hours of incubation, they were subjected to co-immunoprecipitation and western blotting as described in the materials and methods section. Western Blot showing the association between TLR-2 and MyD88. The blot was reprobed and blotted with anti β-actin antibody to confirm equal protein loading. The western blot is the most representable of three independent experiments each showing similar results.
Effect of Ara-LAM on IRAK-1-MyD88 association, induction of IRAK-1 activity and IRAK-M expression in Man-LAM treated macrophages
TLR-2 and MyD88 association initiates a chain of signaling cascades involving the recruitment of different kinases, namely IRAK-1 and IRAK-4. IRAKs [44, 45], including IRAK-4, recruits IRAK-1 association with MyD88, leading to activation and phosphorylation of IRAK-1. This step is essential for phosphorylation and activation of Ikk-α [44, 45]. Hence our previous results prompted us to study the effect of Ara-LAM pre-treatment on IRAK-1 association with MyD88, activation of IRAK-1 and expression of IRAK-M. We performed co-immunoprecipitation of the cell lysates with anti-IRAK-1 antibody and probed the blot with anti-MyD88 antibody. We observed a distinct association between IRAK-1 and MyD88 in Ara-LAM pretreatment in both untreated and Man-LAM treated macrophages (Fig. 3a, lanes 2 and 4) Not only that, we further studied the enzymatic activity of IRAK-1 by performing the in vitro kinase assay using MBP as substrate, where we found that Ara-LAM pre-treatment of macrophages exhibited a significant upregulation of IRAK-1 activity both in untreated or Man-LAM treated macrophages (Fig. 3d, lanes 2 and 4). In addition to this we have also studied the expression of IRAK-1 and found that Ara-LAM pretreatment significantly increases the expression of IRAK-1 both in untreated and Man-LAM treated macrophages (Fig. 3c, lanes 2 and 4).
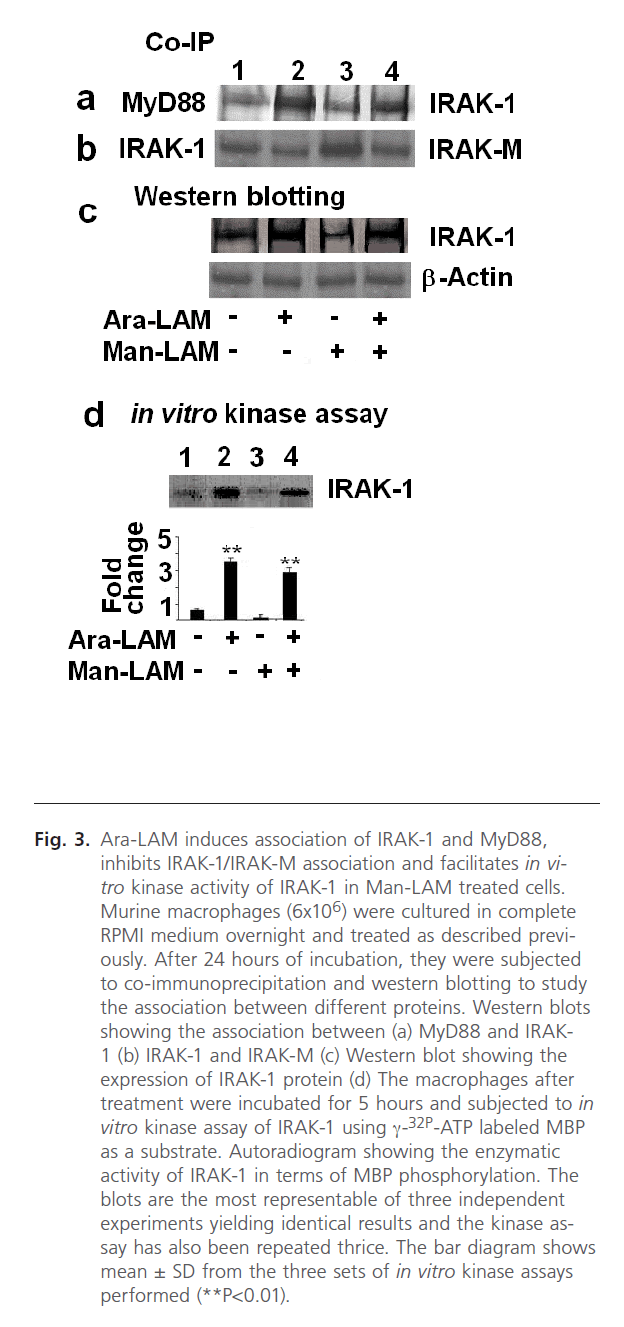
Fig 3: Ara-LAM induces association of IRAK-1 and MyD88, inhibits IRAK-1/IRAK-M association and facilitates in vitro kinase activity of IRAK-1 in Man-LAM treated cells. Murine macrophages (6x106) were cultured in complete RPMI medium overnight and treated as described previously. After 24 hours of incubation, they were subjected to co-immunoprecipitation and western blotting to study the association between different proteins. Western blots showing the association between (a) MyD88 and IRAK- 1 (b) IRAK-1 and IRAK-M (c) Western blot showing the expression of IRAK-1 protein (d) The macrophages after treatment were incubated for 5 hours and subjected to in vitro kinase assay of IRAK-1 using γ-32P-ATP labeled MBP as a substrate. Autoradiogram showing the enzymatic activity of IRAK-1 in terms of MBP phosphorylation. The blots are the most representable of three independent experiments yielding identical results and the kinase assay has also been repeated thrice. The bar diagram shows mean ± SD from the three sets of in vitro kinase assays performed (**P< 0.01).
The above results encouraged us to focus on the negative regulation of TLR-2 signaling mediated by IRAK-M. Man-LAM has been reported to dampen the TLR-2 mediated signaling by induction of the IRAK-M [23]. Therefore, we performed co-immunoprecipitation with IRAK-M and IRAK-1 to study the association between these two proteins, and observed that Man-LAM induced IRAK-M-IRAK-1 association could be significantly inhibited by Ara-LAM pretreatment of either untreated or Man-LAM treated macrophages (Fig. 3b, lanes 2 and 4), showing that Ara-LAM could facilitate TLR-2 mediated signaling in Man-LAM treated cells by inhibiting the negative regulation of IRAK-M.
Effect of Ara-LAM on Ikk-α and IkB-α expression in Man-LAM treated macrophages
The activation of IRAKs facilitates the downstream signaling of TLRs, and the phosphorylation of different accessory proteins. Amongst these, Ikk-α is a very important protein that catalyses the phosphorylation and degradation of IkB-α [45]. Hence we concentrated on the study of these two proteins. Western blot was performed to study the expression and in vitro kinase assay to study Ikk-α activity. Our data clearly showed that Ara-LAM pre-treatment of either untreated or Man-LAM treated macrophages significantly enhanced the activity of Ikk-α (Fig. 4a, lanes 2 and 4). Besides, we also observed a considerable abrogation in the IkB-α expression in Ara-LAM pretreated cells compared to that in Man-LAM treated cells (Fig. 4b, lanes 2 and 4).
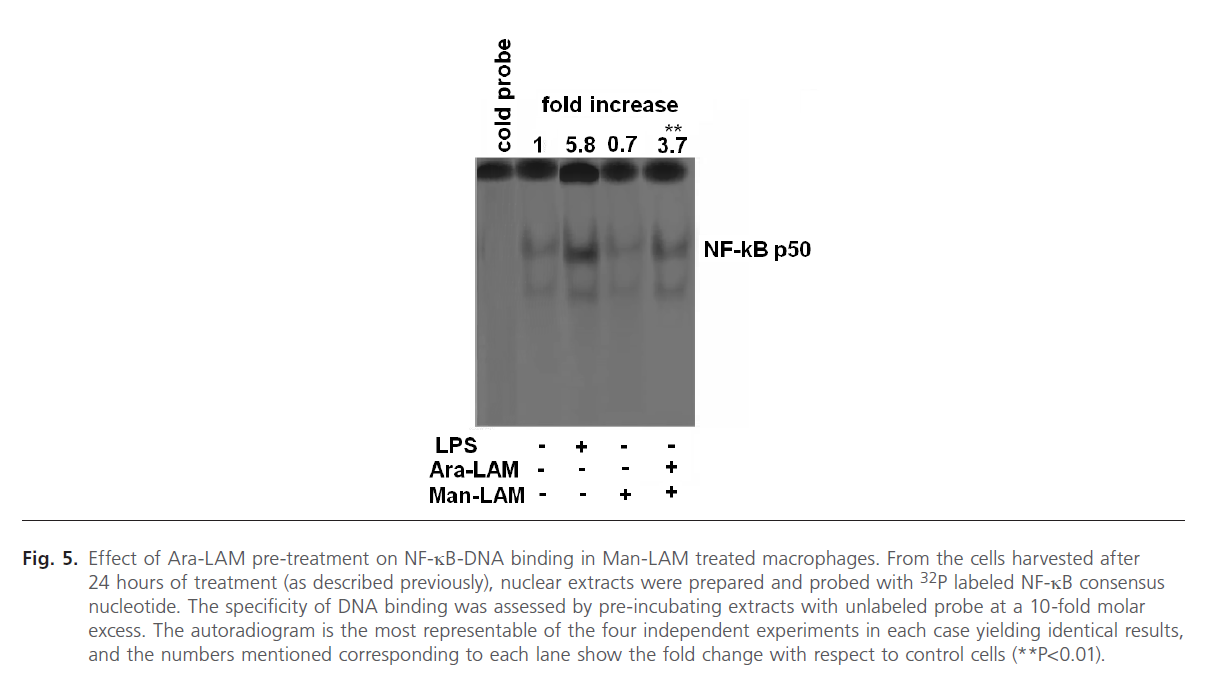
Fig 4: Ara-LAM enhances IkK kinase activity and IkB-α degradation in Man-LAM-treated macrophages. Murine peritoneal macrophages (6x106) cultured overnight as described previously were subjected to Ara-LAM treatment at a dose of 3 µg/ml for 3 hours, washed, followed by Man-LAM treatment (10 µg/ml). The cells were incubated further for 5 hours for studying the IkK kinase activity and 24 hours for studying the IkB-α degradation by western blotting. (a) Autoradigram showing the enzymatic activity of IkK (b) Western blot showing the expression of IkB-α. The blot was reprobed and blotted with anti-β actin antibody to confirm equal protein loading. Both the autoradiogram and blot are the most representable of three independent experiments showing the similar effects. The bar diagram for the in vitro kinase assay represent mean ± SD of the three experiments (*P< 0.05, **P< 0.01).
Effect of Ara-LAM pre-treatment on NF-kB-DNAbinding in Man-LAM treated macrophages
Since the degradation of IkB-a from the NF-kB-IkB complex leads to translocation of the free NF-kB to the nucleus and DNA binding [46, 47], we further studied the effect of Ara- LAM pre-treatment on nuclear translocation of NF-kB by EMSA assay of nuclear protein extracts of Man-LAM treated cells. We also treated the cells with LPS (1 mg/ml) as a positive control which is a known inducer of NF-kB tranlocation and DNA binding (Fig. 5, lane 3). We observed that Man-LAM treatment caused a down regulation of NF-kB translocation in naïve macrophages (Fig. 5, lane 4), but Ara-LAM pretreatment of murine peritoneal macrophages followed Man-LAM treatment exhibited a significant enhancement of NF-kB binding activity compared to that of untreated control (Fig. 5, lane 5). Densitometric analysis of the NF-kB specific bands indicated that there was about a 5 fold increase in DNA binding in Ara-LAM pre-treated cells.

Fig 5: Effect of Ara-LAM pre-treatment on NF-kB-DNA binding in Man-LAM treated macrophages. From the cells harvested after 24 hours of treatment (as described previously), nuclear extracts were prepared and probed with 32P labeled NF-kB consensus nucleotide. The specificity of DNA binding was assessed by pre-incubating extracts with unlabeled probe at a 10-fold molar excess. The autoradiogram is the most representable of the four independent experiments in each case yielding identical results, and the numbers mentioned corresponding to each lane show the fold change with respect to control cells (**P< 0.01).
Ara-LAM mediated protection in Man-LAM induced pathogenesis is TLR-2 dependent
Finally we were interested to investigate whether; silencing of TLR-2 gene could impair the Ara-LAM mediated protection against Man-LAM induced pathogenesis. Initially we standardized the dose and the silencing/blocking action of the TLR-2-siRNA and the anti TLR-2 antibody (Fig. 6a). We transfected the murine macrophages with TLR-2 specific siRNA (100 ng/ml) for 24 hours in a low FBS medium (as described in the methods section), or incubated with anti-TLR-2 Ab (4 mg/ml) for 2 hours, after which Ara-LAM pretreatment and Man-LAM treatments were given. We performed RT-PCR to study the expression of TNF-α, IL-12 p40 expression and iNOS mRNA expression and also studied the nitrite generation from the cell free supernatants of sets incubated up to 48 hrs of treatment. It was clearly observed that silencing of TLR-2 with either TLR-2 specific siRNA, or blocking TLR-2 signaling by anti TLR-2 Ab distinctly abrogated the pro-inflammatory cytokine and iNOS expression (Fig. 6 b-d, lanes 5 and 6) as well as NO generation (Fig. 6f) in untreated macrophages or Man-LAM treated macrophages under Ara-LAM pretreated condition.
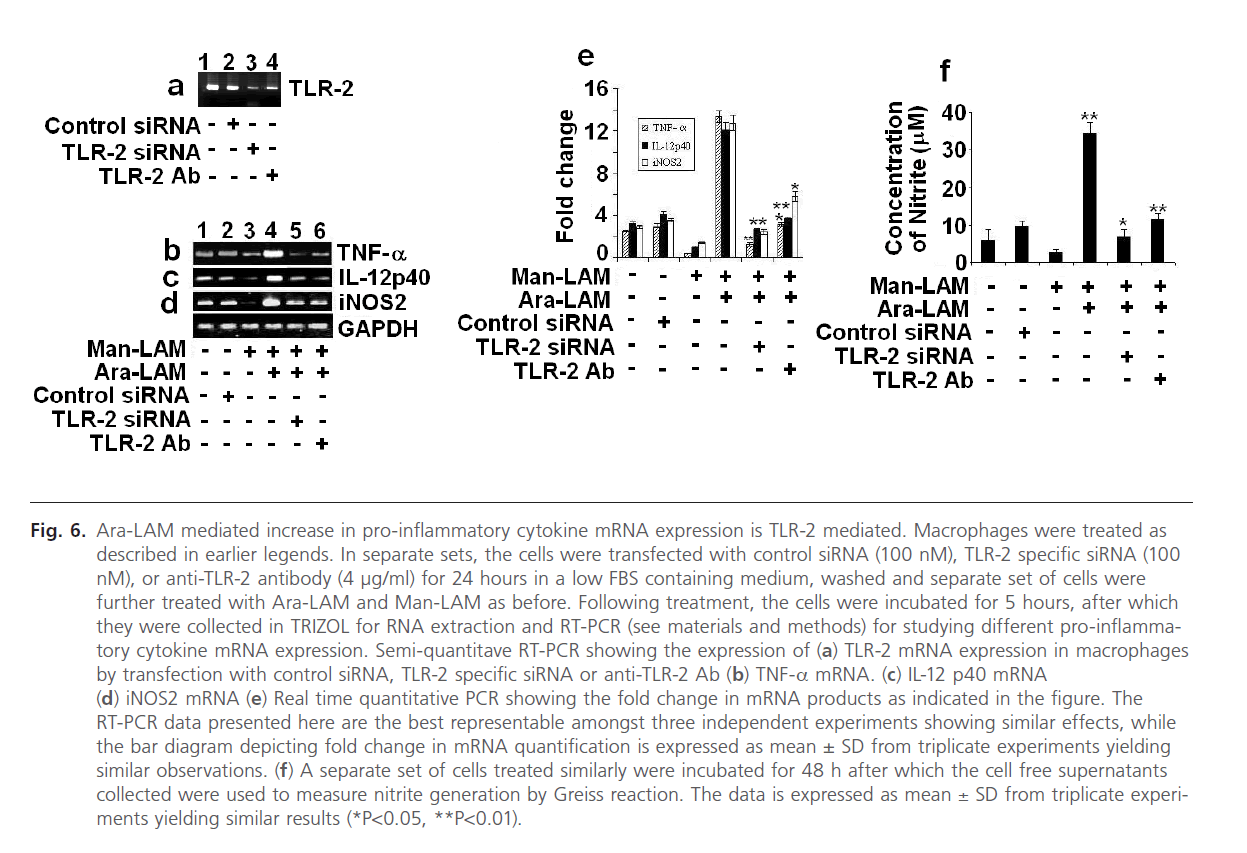
Fig 6: Ara-LAM mediated increase in pro-inflammatory cytokine mRNA expression is TLR-2 mediated. Macrophages were treated as described in earlier legends. In separate sets, the cells were transfected with control siRNA (100 nM), TLR-2 specific siRNA (100 nM), or anti-TLR-2 antibody (4 μg/ml) for 24 hours in a low FBS containing medium, washed and separate set of cells were further treated with Ara-LAM and Man-LAM as before. Following treatment, the cells were incubated for 5 hours, after which they were collected in TRIZOL for RNA extraction and RT-PCR (see materials and methods) for studying different pro-inflammatory cytokine mRNA expression. Semi-quantitave RT-PCR showing the expression of (a) TLR-2 mRNA expression in macrophages by transfection with control siRNA, TLR-2 specific siRNA or anti-TLR-2 Ab (b) TNF-a mRNA. (c) IL-12 p40 mRNA (d) iNOS2 mRNA (e) Real time quantitative PCR showing the fold change in mRNA products as indicated in the figure. The RT-PCR data presented here are the best representable amongst three independent experiments showing similar effects, while the bar diagram depicting fold change in mRNA quantification is expressed as mean ± SD from triplicate experiments yielding similar observations. (f) A separate set of cells treated similarly were incubated for 48 h after which the cell free supernatants collected were used to measure nitrite generation by Greiss reaction. The data is expressed as mean ± SD from triplicate experiments yielding similar results (*P< 0.05, **P< 0.01).
Discussion
Protection against infectious diseases is regulated by the interplay of cytokines with the cell-mediated immune responses. The major virulence factor involved in the induction of pro-pathogenic Th2 responses is reported to be a cell wall glycolipid called Mannosylated Lipoarabinomannan (Man-LAM). On the contrary, Ara-LAM produced by nonpathogenic mycobacteria is a potent inducer of both proinflammatory cytokines and the anti-inflammatory cytokine IL-10. However, from our previous study we established that Ara-LAM induced IL-10 possesses a pro-inflammatory function [32] not only that, Ara-LAM also restores the impaired cell mediated immune responses in a murine model of tuberculosis [48]. We have, therefore, undertaken this study to understand the upstream mechanism of the Ara-LAM mediated protection during mycobacterial pathogenesis.
Ara-LAM is known to have immunomodulatory properties, the most important of them being the induction of Toll-Like Receptor-2 (TLR-2) mediated transcription of pro-inflammatory cytokines [21, 22]. Since TLR-2 plays a pivotal role in innate immune response and induces adaptive immunity, we studied its expression in Ara-LAM pretreated cells, followed by treatment with Man-LAM. It was clearly observed that TLR-2 expression on the macrophage surface (Fig. 1) was significantly enhanced in Man-LAM treated or untreated cells under Ara-LAM pretreatment at a noncytotoxic dose of 3 mg/ml. The TLR-2 signaling is initiated by the association of TLR-2 and MyD88, an adapter protein [44], and their association triggers the downstream cascade, leading to the activation of IRAKs. These kinases catalyze the phosphorylation of a series of accessory proteins like NIKs, Ikk and the series of downstream signaling ultimately culminating in the translocation of NF-kB to the nucleus [44-46]. As per our findings, we also observed a strong selective association between TLR-2 and MyD88 (Fig. 2) in Ara-LAM pretreated cells with or without Man-LAM treatment. Probing the downstream signaling, Ara-LAM pretreatment of untreated and Man-LAM treated macrophages exhibited a significant induction of IRAK-1 expression and in vitro kinase activity, along with inhibition of IRAK-M/IRAK-1 association. IRAK-M is a negative regulator of TLR-2 mediated signaling, and Man-LAM has been reported to up-regulate IRAK-M expression, thus dampening the TLR-2 mediated upregulation of pro-inflammatory cytokine mRNA transcription [23]. Since Ara-LAM treatment inhibits IRAKM expression, it is likely that this compound facilitated the progression of downstream signaling cascade in Man-LAM treated cells as well.
Tracing further downstream of the TLR-2 signaling pathway, we observed that Ara-LAM pretreatment induced Ikk-α expression and enzymatic activity which led to the phosphorylation and degradation of IkB-α from the NF-kB-IkB complex in the cytosol (Fig. 4) in untreated and Man-LAM treated cells. Subsequently, there is a significant translocation and DNA binding of NF-kB in Ara-LAM pretreated cells as shown by EMSA (Fig. 5). The expression of pro-inflammatory cytokines and Nitric Oxide (NO) is regulated by the different transcription factors like NF-kB [47]. Earlier reports established that NF-kB translocation to the nucleus and its binding to DNA are down-regulated by Man-LAM, but Ara-LAM facilitates NFkB- DNA binding [49]. It is also known that Toll-Like Receptor- 2 (TLR-2) activation induces NF-kB dependent induction of pro-inflammatory cytokine transcription [47, 49]. Finally it was observed that the induction of pro-inflammatory cytokines TNF-α and IL-12p40, as well as iNOS mRNA by Ara-LAM is TLR-2 dependent, as confirmed by transfection experiments with TLR-2 specific siRNA (Fig. 6).
Thus from our detailed investigations involving the mechanism of Ara-LAM mediated protection during Man-LAM induced pathogenesis, we suggest that this glycolipid antigen isolated from non-pathogenic mycobacteria could be of immense importance in the field of anti-tuberculosis immunotherapy, since it could induce the pro-inflammatory cytokines creating an anti-pathogenic environment within the host cell. This is a first time report that unveils the mechanism of Ara-LAM mediated protection against mycobacterial pathogenesis by triggering TLR-2 mediated signal transduction in Man-LAM treated cells, thus hinting at TLR-2 signaling as a therapeutic target in anti-mycobacterial immunotherapy.
Acknowledgements
This work has been funded by Council of Scientific and Industrial Research (CSIR), Govt. of India. We express our gratitude to the Director, Bose Institute for his support. Special thanks are expressed to Prof. D.J. Chattopadhyay, Dean of Science, University of Calcutta, for helping us with Real Time PCR experiments. We also thank Dr. Alok K. Sil, Dept. of Microbiology, University College of Science, for helping us with the in vitro kinase assays. Mr. Prabal Gupta is acknowledged for his technical assistance.
1854
References
- Saunders, B.M., Frank, A.A., Orme, I.M. Granuloma formation is required to contain bacillus growth and delay mortality in mice chronically infected with Mycobacterium tuberculosis. Immunology 1999; 98: 324-8.
- Cardona, P.J, Llatjos, R., Gordillo, S., Diaz, J., Ojanguren, I., Ariza, A., Austina, V. Evolution of granulomas in lungs of mice infected aerogenically with Mycobacterium tuberculosis. Scand. J. Immunol. 2000; 52: 156-163.
- Wallis, R.S., Amir-Tahmasseb, M., Ellner, J.J. Induction of IL-1 and TNF-alpha by mycobacterial proteins: the monocyte western blot. PNAS USA 1990; 87: 3348-3352.
- Riedel, D.D., Kaufmann, S.H.E. Chemokine secretion by human polymorphonuclear granulocytes after stimulation with Mycobacterium tuberculosis and LAM. Infect. Immun. 1997; 65: 4620-4623.
- Sibley, L.D., Hunter, S.W., Brennan, P.J., Krahenbuhl, J.L. Mycobacterial Lipoarabinomannan inhibits gamma-interferon-mediated activation of Macrophages. Infect. Immun. 1998; 56: 1232 1236.
- Wilker, H.G. Liberation of soluble proteins from live and dead mycobacterial cells and the implications of pathogenicity of tubercle bacilli hypothesis. Scand. J Immunol. 2001; 54: 82-6.
- Chan, J., Fan, X., Hunter, S.W., Brennan, P.J., Bloom, B.R. Lipoarabinomannan, a possible virulence factor involved in the persistence of Mycobacterium tuberculosis within macrophages. Infect. Immun. 1991; 59: 1755-1761.
- Hunter, S.W., Gaylord, H., Brennan, P.J. Structure and antigenicity of the phosphorylated lipopolysaccharide antigens from leprosy and tubercle bacilli. J Biol. Chem. 1986; 261: 12345-12351.
- Hunter, S.W., Brennan, P.J. Evidence for the presence of a phosphatidyl inositol anchor on Lipoarabinomannan and Lipomannan of Mycobacterium tuberculosis. J Biol. Chem. 1991; 265: 9272-9279.
- Chatterjee, D., Khoo, K.H. Mycobacterial Lipoarabinomannan: an extraordinary lipoheteroglycan with profound physiological effects. Glycobiology 1998; 8: 113-120.
- Kaplan, G., Gandhi, R.R., Weinstein, D.E., Levis, W.R., Patarroyo, M.E., Brennan, P.J., Cohn, Z.A. Mycobacterium leprae antigen induced suppression of T-cell proliferation in vitro. J. Immunol. 1987; 138: 3028-3034.
- Rojas, M., Garcia, L.F., Nigou, J., Puzo, G., Olivier, M. Mannosylated Lipoarabinomannan antagonizes Mycobacterium tuberculosis induced macrophage apoptosis by altering Ca+2 dependent cell signaling. J. Infect. Dis. 2000; 182: 240-251.
- Maiti, D., Bhattacharyya, A., Basu, J. Lipoarabinomannan from M. tuberculosis promotes macrophage survival by phosphorylation of Bad through PI3K-Akt pathway. J. Biol. Chem. 2001; 276: 329-333.
- Sirkar, M., Majumdar, S. Lipoarabinomannan induced cell signaling involves Ceramide and mitogen activated protein kinase. Clin Diagn Lab Immunol. 2002; 9: 1175-182.
- Ghosh, S., Das, S., Dasgupta, S.K., Majumdar, S. Lipoarabinomannan induced cytotoxic effects in human mononuclear cells, FEMS Immunol. Med. Microbiol. 1998; 21: 181-188.
- Juffermans, N.P., Verbon, A., Belisle, J.T., Hill, P.J., Speelman, P., van Deventer, S.J., van der Poll, T. Mycobacterial lipoarabinomannan induces an inflammatory response in the mouse lung. A role for interleukin-1. Am. J. Respir. Crit. Care Med. 2000; 162: 486-489.
- Roach, T.I., Barton, C.H., Chatterjee, D., Blackwell, J.M. Macrophage activation: lipoarabinomannan from avirulent and virulent strains of Mycobacterium tuberculosis differentially induces the early genes c-fos, KC, JE and tumor necrosis factor-a. J. Immunol. 1993; 150: 1886-1896.
- Zhang, Y., Doerfler, T., Lee, C., Guillemin, B., Rom, W.N. Mechanisms of stimulation of Interleukin-1b and tumor necrosis factor-a by Mycobacterium tuberculosis components. J. Clin. Invest. 1993; 91: 2076-2083.
- Roach, T.I., Barton, C.H., Chatterjee, D., Liew, F.Y., Blackwell, J.M. Opposing effects of interferon-g on iNOS and interleukin-10 expression in lipopolysaccharide-and mycobacterial lipoarabinomannan-stimulated macrophages. Immunology 1995; 85: 106-113.
- Means, T.K., Wang, S., Lien, E., Yoshimura, A., Golenbock, D.T., Fenton, M.J. Human toll like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 1999; 7: 3920-3927.
- Wieland, C.W., Knapp, S., Florquin, S., de Vos, A.F., Takeda, K., Akira, S., Golenbock, D.T., Verbon, A., van der Poll, T. Non-mannose-capped lipoarabinomannan induces lung inflammation via Toll-like receptor 2. Am. J. Crit. Care Med. 2004; 12: 1367-1374.
- Iwasaki, A., Medzhitov, R. Toll like receptor control activation of the adaptive immune responses. Nat. Immunol. 2004; 5: 971-974.
- Pathak, S.K., Basu, S., Bhattacharyya, A., Pathak, S., Kundu, M., Basu, J. Mycobacterium tuberculosis lipoarabinomannan-mediated IRAK-M induction negatively regulates Toll-like receptor-dependent interleukin-12 p40 production in macrophages. J. Biol. Chem. 2005; 280: 42794-427800.
- Dahl, K.E., Shiratsuchi, H., Hamilton, B.D., Ellner, J.J., Toossi, Z. Selective induction of Transforming Growth Factor beta in human monocytes by lipoarabinomannan of Mycobacterium tuberculosis. Infect. Immun. 1996; 64: 399-405.
- Gazzinelli, R.T., Oswald, I.P., James, S.L., Sher, A. IL-10 inhibits parasite killing and nitric oxide production by IFN-g activated macrophages. J. Immunol. 1992; 148: 1792-1796.
- Murray, P.J., Wang, L., Onufryk, C., Tepper, R.I., Young, R.A. T-cell derived IL-10 antagonises macrophage function in mycobacterial infection. J. Immunol. 1997; 158: 315-321.
- Rojas, M., Olivier, M., Gros, P., Barrera, L.F., Garcia, L.F. TNF Alpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J. Immunol. 1999; 162: 6122- 6131.
- Bhattacharjee, S., Majumder, N., Bhattacharyya, P., Bhattacharyya, S., Majumdar, S. Immunomodulatory role of arabinosylated lipoarabinomannan on Leishmania donovani infected murine macrophages. Ind. J. Biochem. Biophys. 2007; 44: 366-372.
- Bhattacharyya, P., Bhattacharjee, S., Gupta, G., Majumder, S., Adhikari, A., Mukherjee, A., Majumdar, S.B., Saha, B., Majumdar, S. Arabinosylated Lipoarabinomannan – mediated protection in visceral leishmaniasis through up-regulation of toll like receptor 2 signaling: an immunoprophylactic approach. J. Inf. Dis. 2010; 202: 145-155.
- Dascher, C.C., Hiromatsu, K., Xiong, X., Morehouse, C., Watts, G., Liu, G., McMurray, D.N., LeClair, K.P., Porcelli, S.A., Brenner, M.B. Immunization with a mycobacterial lipid vaccine improves pulmonary pathology in the guinea pig model of tuberculosis. Int. Immunol. 2003; 15: 915-925.
- Hamasur, B., Haile, M., Pawlowski, A., Schroder, U., Williams, A., Hatch, G., Hall, G., Marsh, P., Kallenius, G., Svenson, S.B. Mycobacterium tuberculosis arabinomannan-protein conjugates protect against tuberculosis. Vaccine 2003; 21: 4081-4093.
- Majumder, N., Dey, R., Mathur, R.K., Datta, S., Maitra, M., Ghosh, S., Saha, B., Majumdar, S. An unusual pro-inflammatory role of interleukin-10 induced by arabinosylated lipoarabinomannan in murine peritoneal macrophages. Glycoconj. J. 2006; 23: 675-686.
- Fahey, T., Tracey, K.J., Olson, P.T., Consens, L.S., Jones, W.G., Shires, G.T., Sherry, B. Macrophage inflammatory protein I modulates macrophage function. J. Immunol. 1992; 148: 2764-2769.
- Denizot, F., Lange, R. Rapid colorimetric assay for cell growth and survival Modification to the tetrazolium dye procedure giving improved sensibility and Reliability. J. Immunol. Meth. 1986; 89: 271- 277.
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. PNAS USA 1979; 76: 4350-4354.
- Majumdar, S., Rossi, M.W., Fujiki, T., Phillips, W.A., Disa, S., Queen, C.F., Johnston R.B., Rosen, O.M., Corkey, B.E., Korchak, H.M. Protein Kinase C isotypes and signaling in neutrophils, J. Biol. Chem. 1991; 266: 9285-9294.
- Ghosh, S., Bhattacharyya, S., Sirkar, M., Sa, G.S., Das, T., Majumdar, D., Roy, S. Majumdar S, Leishmania donovani suppresses activated protein 1 and NF-kB in host macrophages via ceramide generation: involvement of extracellular signal regulated kinase. Infect. Immun. 2002; 70: 6828-6838.
- Maniatis, T., Fritsch, E.F., Sambrook, J. Molecular cloning: a laboratory Manual, Cold Spring Harbor, NY: Cold Spring Harbor laboratory Press. 1989.
- Chomezynaski, P., Sacchi, N. Single step method for RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987; 162: 156-159.
- Green, L.C., Wanger, D.A., Glogowski, J., Skipper, P.L., Wishnok, J.S., Tannenbaum, S.R. Analysis of nitrate, nitrite and nitrate in biological fluid. Anal. Biochem. 1982; 126: 131-138.
- Hibbs, J.B., Taintor, R., Vavrin, Z., Rachlin, E. Nitric Oxide: a cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988; 157: 87-94.
- Cooper, A.M., Kipnis, A., Turner, J., Magram, J., Ferrante, J., Orme, I.M. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J. Immunol. 2002; 168: 1322-1327.
- Takeda, K., Akira, S., Kaisho, T. Toll like Receptors: Critical Proteins linking innate and acquired immunity. Nat. Immunol. 2001; 2: 675- 680.
- Li, S., Strelow, A., Fontana, E.J., Wesche, H. IRAK-4: A novel member of the IRAK family with the property of an IRAK-kinase. Proc. Nat. Atl. Sc. USA 2002; 99: 5567-5572.
- Takeda, K., Akira, S. Toll like Receptor Signalling, Nat. Immunol. 2004; 4: 499-511.
- Baldwin, A.S. The NF-kB and IkB proteins: new discoveries and insights, Annu. Rev. Immunol. 1996; 14: 649-683.
- Ukil, A., Biswas, A., Das, T., Das, P.K. 18b-Glycyrrhetinic acid triggers curative Th1 response and nitric oxide up-regulation in experimental visceral leishmaniasis associated with the activation of NF-kB. J. Immunol. 2005; 175: 1161-1169.
- Majumder, N., Bhattacharjee, S., Dey, R., Bhattacharyya Majumdar, S., Pal, N.K., Majumdar, S. Arabinosylated Lipoarabinomannan modulates the impaired cell mediated immune response in Mycobacterium tuberculosis H37Rv infected C57BL/6 mice. Microbes. Infect. 2008; 10: 349-357.
- Brown, M.C., Taffet, S.M. Lipoarabinomannan derived from different strains of Mycobacterium tuberculosis differentially stimulate the activation of NF-kappa B and KBF1 in murine macrophages. Infect. Immun. 1995; 63: 1960-1968.