Chenguang Li, Songjie Liao and Jian Yu*
Department of Neurology, Sun Yat-sen University, Guangzhou 510080, PR China
*Corresponding Author:
Jian Yu
Department of Neurology
Guangdong Key Laboratory for Diagnosis
and Treatment of Major Neurological Diseases
National Key Clinical Department
National Key Discipline
the First Affiliated Hospital of Sun Yat-sen University
Guangzhou 510080, PR China
Tel: +86-2087755766-8291
Fax: +86-2087606221
E-mail: yujian21cn@163.com
Citation: Li C, Liao S, Yu J. Tuberous Sclerosis Complex Confirmed by Genetic Analysis: A Case Report. J Neurol Neurosci. 2016, 6:4. doi: 10.21767/2171-6625.100046
Received Date: November 27, 2015; Accepted Date: November 30; 2015, Published Date: December 04, 2015
Tuberous sclerosis complex (TSC), also known as Bourneville disease, is an autosomal-dominant, neurocutaneous, multisystem disorder [1]. The underlying genetic cause is mutations in the TSC1 or TSC2 gene, which leads to overactivation of the mammalian target of rapamycin (mTOR) protein complex [2,3]. The main clinical features of the disease are facial sebaceous adenomas, epilepsy, and hypophrenia. Currently diagnosis is based on the criteria established at the 2012 International Tuberous Sclerosis Complex Consensus Conference [4]. mTOR inhibition is presently the main treatment [2,5]. It is still difficult to differentially diagnose TSC from other conditions such as neurofibromatosis, epilepsy, and mental disease, which makes genetic analysis important in its diagnosis.
Keywords
Tuberous sclerosis complex; Genetic analysis; Case report
Introduction
Tuberous sclerosis complex (TSC), also known as Bourneville disease, is an autosomal-dominant, neurocutaneous, multisystem disorder [1]. The underlying genetic cause is mutations in the TSC1 or TSC2 gene, which leads to overactivation of the mammalian target of rapamycin (mTOR) protein complex [2,3]. The main clinical features of the disease are facial sebaceous adenomas, epilepsy, and hypophrenia. Currently diagnosis is based on the criteria established at the 2012 International Tuberous Sclerosis Complex Consensus Conference [4]. mTOR inhibition is presently the main treatment [2,5]. It is still difficult to differentially diagnose TSC from other conditions such as neurofibromatosis, epilepsy, and mental disease, which makes genetic analysis important in its diagnosis.
Case Report
We report the case of patient with TSC associated with a novel mutation in the TSC2 gene. In May 2015, a 31-year-old man was admitted to our hospital with a 7-months history of sudden-onset loss of consciousness, characterized by lack of responsiveness to any external stimuli, and sometimes accompanied by urinary incontinence. Each attack lasted for 4-5 seconds, with a frequency of 1-2 episodes a day. The patient was unaware of the attack, and often felt dizziness before onset. He visited our outpatient clinic, and was diagnosed with secondary epilepsy. After treated with oxcarbazepine for a week, he did not experience any further attacks, His previous medical history, personal history, and family history revealed no significant findings. Physical examination revealed many sebaceous adenomas on the face. Results of a neurological examination were normal.
MRI showed multiple patchy signal abnormalities in the brain, and round abnormal signal intensity deep in the left temporal lobe (Figure 1). CT revealed similar abnormalities (Figure 2). Significant abnormal recordings were observed on long-term video EEG. Abdominal Doppler ultrasound showed multiple hyperechoic tubers in the liver. Results of a CSF examination were normal with the exception of mild elevation of IgA, IgM, and IgG.
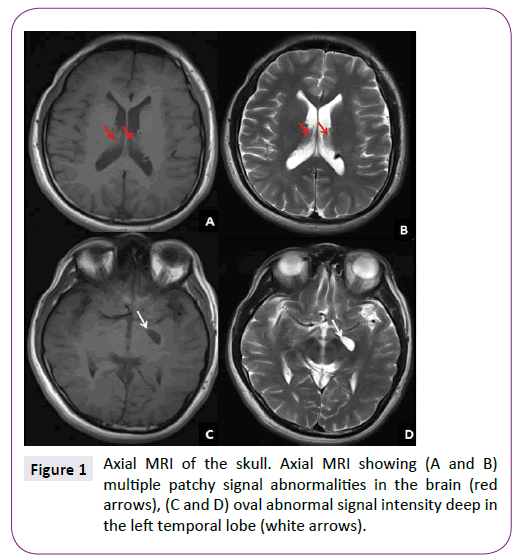
Figure 1: Axial MRI of the skull. Axial MRI showing (A and B) multiple patchy signal abnormalities in the brain (red arrows), (C and D) oval abnormal signal intensity deep in the left temporal lobe (white arrows).
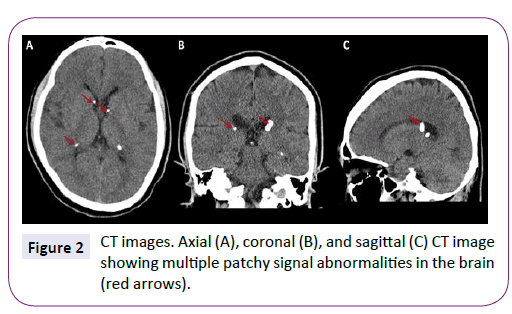
Figure 2: CT images. Axial (A), coronal (B), and sagittal (C) CT image showing multiple patchy signal abnormalities in the brain (red arrows).
Genetic analysis of a blood sample revealed a novel mutation in the TSC2 gene (TSC2 c.3355C>T p. (Gln1119*) heterozygosis). Based on these findings, the patient was diagnosed with TSC.
Discussion
According to the diagnostic criteria mentioned previously, this case displayed two major features and one minor feature. Therefore, it can be classified as a “definite diagnosis,” which was confirmed by genetic testing. Although this patient visited our hospital for epilepsy treatment, we detected a novel mutation in the TSC2 gene. Therefore, it is necessary for doctors to be familiar with the clinical features of TSC to confirm the diagnosis with genetic testing if possible.
Acknowledgments
This work was supported by the project of National Natural Science Foundation of China (No.81371276) and Natural Science Fund of Guangdong (No.2014A030313065) to Jian Yu.
7889
References
- DiMario FJ, Sahin M, Ebrahimi-Fakhari D (2015) Tuberous Sclerosis Complex. PediatrClin North Am 62:633-648.
- Curatolo P (2015) Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. PediatrNeurol 52:281-289.
- Wataya-Kaneda M (2015) Mammalian target of rapamycin and tuberous sclerosis complex. J DermatolSci 79:93-100.
- Northrup H, Krueger DA (2013) Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. PediatrNeurol 49:243-54.
- Tran LH, Zupanc ML (2015) Long-Term Everolimus Treatment in Individuals with Tuberous Sclerosis Complex: A Review of the Current Literature. PediatrNeurol 53:23-30.

