Keywords
|
| Chalcone; Docking; Plasmodium falciparum |
Introduction
|
| Malaria causes 1 million deaths every year worldwide among about 250 million people suffering from this major parasitic infection, according to the World Health Organization [1]. There are four species of plasmodium namely P. falcipurum, P. ovale, P. vivax and P. malariae. In above mentioned, only P. falcipurum accounts for over 95% of all malaria cases mainly in Africa and affects more children under five years of age [2,3]. Classical anti-malarial agents such as chloroquine, mefloquine, and anti-folates, have prompted researchers world-wide to consider and to search for other antimalarial drugs of ideally different molecular mechanism of action [4]. The increasing of resistant strains to chloroquine has raised the search of new potential drugs [5] and the artemisin like substances are promising candidates in order to control this epidemic and intensive research is being made on cyclic-peroxy compounds [5]. Artemisinins, if deployed indiscriminately as monotherapy, will soon fall to resistance. This led World Health Organization (WHO) to advocate the use of artemisinins in combination with another anti-malarial so that emergence of resistance to this vital class of drug could be postponed. Combination therapy also decreases pressure on the rising demand of artemisinin, as it is a natural product in short supply [5,6], thus it is necessary to search an alternate drug. |
| A small number of chemically related drugs belonging to five classes of compounds: 4-aminoquinolines (chloroquine (CQ), quinine, mefloquine, amodiaquine and halofantrine), 8-aminoquinolines (primaquine), anti-folate compounds, atovaquone and artemisinin derivatives are currently in use [7]. Naturally occurring chalcones are the key intermediates for various plant metabolites. They are biologically active compounds with known antibacterial [8,9], antifilarial [10], antiviral [11,12], anti-leishmanial [13] and cytotoxic [14,15] activities. Hydroxychalcones, as an important part of chalcones family, are widespread among natural and synthesized chalcones and have attracted an increased attention [16-18]. Therefore it is necessary to search the biologically potent derivatives. Herein we consider the theoretical root for search. Quantitative structure-activity relationship (QSAR) methods are used extensively in the design and development of new antimalarial compounds [19-21]. Docking procedures permits virtually screening a database of compounds and predict the strongest binder on various scoring functions. It finds ways in which two molecules, such as drugs and an enzyme and/or protein fit together and dock to each other well [22]. Molecular docking is used to study how a ligand is interacting with its biological target and to further support the conclusions of QSAR studies [23-29]. |
| Falcipain-2 and falcipain-3 are critical hemoglobinases of Plasmodium falciparum, the most virulent human malaria parasite. In this article, we have studied the QSAR and docking studies the previously synthesized chalcones using iGEMDock, a computational tool. |
Experimental
|
|
Compound selection and ligand preparation
|
| All the compounds (52 compounds) were drawn on ChemDraw as in Table 1 and were optimized using MOPAC (a computational tool) suing AM1 calculation and closed shell restricted with RMS gradient 0.1. |
| Protein preparation: Bioinformatics is seen as an emerging field with the potential to significantly improve how drugs are found, brought to the clinical trials and eventually released to the marketplace. The crystal structure of P. falcipurum protease having PDB ID: 1MVT was taken (https://www.rcsb.org/). The protein was prepared using Molegro Molecular Viewer 2.5 and the same was used for the virtual drug screening. |
| Docking studies: Herein, iGEMDock was used for the drug screening as in Table 2. iGEMDock is a molecular docking tool and generates diversity of chalcones conformations from different seeds with high temperature molecular dynamics. Then, it orients the chalcones conformations within the defined protein active site by translating the center of the surfactant. Each orientation is subjected to simulated annealing molecular dynamics and sorted according to the interaction energy. iGEMDOCK energy function consists of electrostatic, steric, and hydrogen- bonding potentials. Steric and hydrogen bonding potentials use a linear model. There are four main steps which are used here. Parameters used for drug screening in iGEMDOCK were as followed: initial step sizes (σ= 0.8 and ψ=0.2), family competition length (L=2), population size (N=200), and recombination probability (pc=0.3). Optimization is set to generate 70 iterations for which it generated 1200 solutions in one generation process and if exceeded then it terminated after 84,000 solutions. |
| Docking studies not only provide an understanding of the binding mode of the ligands but are also employed to validate homology models. The molecular models for P. falciparum were generated by Molegro Molecular Viewer 2.5 docking module. Among the docked poses for each ligand, one with the highest dock score was chosen as the final conformations. Docking allows screening a database of compounds and calculating the strongest binders based on various scoring functions. It explores ways in which two molecules, such as drugs and an enzyme, fit together and docks to each other well. The molecule may bind to receptor and modify their function. The interaction of drug and receptor complex was identified via docking and their relative stabilities were evaluated using molecular dynamics and also evaluated their binding affinities using free energy simulations. |
Results and Discussion
|
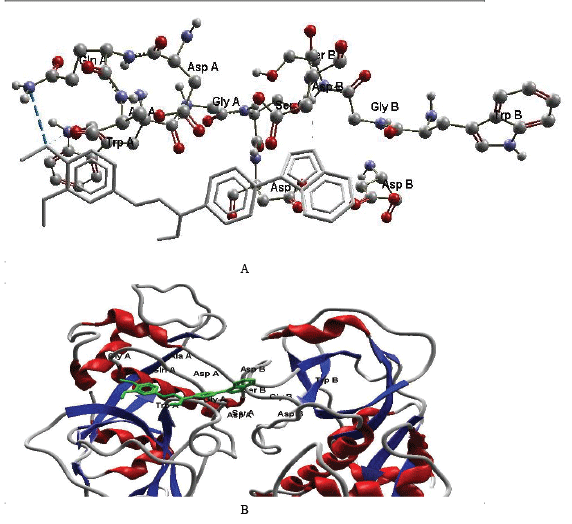
| Docking studies are used extensively in drug discovery such as in the prediction of ligand-receptor complex structures and also to rank the ligand molecules based upon the binding energies of the corresponding ligand-enzyme complex. The objective of our docking study is to elucidate the potential interaction mode of the chalcones with falciparum. A general conclusion derived from these docking results is that the side chain of the ASP and TYR forms hydrogen bonding with the chalcone, 49 as in Figure 1 and details mentioned in Table 2. The nature of these hydrogen bonds involves an interaction between the amidic nitrogen of falcipurum and chalcones. Interestingly, this interaction is almost conserved with all studied inhibitors. The docking calculations provided us with a general static picture of the most energetically favourable binding orientation of inhibitors to the enzyme. To obtain further insight into the dynamic changes of the docked inhibitors within the enzyme active site pocket over time, the lowest energy docked complex of the most active inhibitor, 49, was subjected to unconstrained MD simulations. The post dynamic monitoring of the overall hydrogen bond networks and the electrostatic interaction between the 49 inhibitor and nearby residue clearly revealed that these inhibitors settle comfortably inside the falcipurum active site and its activity was found comparable to standard drug Lichochalone A and the logP value of 49 and Lichochalone A are found similar. |
Conclusion
|
| The continued development of novel antimalarial chemotherapies, particularly those aimed at new pathways, is necessary for the successful treatment of malaria as resistance to presently utilized drugs becomes more widespread. To address this need, we have focused our drug discovery efforts on pfDHOD, the rate-limiting enzyme in the de novo pyrimidine biosynthetic pathway. Unlike its human host, the malaria parasite does not possess a salvage pathway for pyrimidines and must rely on de novo biosynthesis for its metabolic needs. pfDHOD is therefore likely to be an essential enzyme, and homologs from other species have previously been shown to be amenable to small molecule inhibition. A total of 52 compounds were identified as sub-micromolar inhibitors of which 33 were specific for pfDHOD as compared with hsDHOD. The low observed hit rate of 0.018% is a consequence of the stringent screening criteria. |
Tables at a glance
|
 |
|
| Table 1 |
Table 2 |
|
| |
Figures at a glance
|
 |
| Figure 1 |
|
| |


